


プレスリリース 「ハンチントン病の新しい治療戦略を開発」―第3の細胞死を標的とする神経変性疾患治療の可能性をひらく―
プレスリリース
国立大学法人東京医科歯科大学
国立研究開発法人日本医療研究開発機構
ポイント
- 主要な神経変性疾患のハンチントン病で新しい細胞死TRIADが起きていることを示しました。
- 新しい細胞死TRIADの細胞内シグナル経路の詳細を解明しました。
- 新しい細胞死TRIADを標的とする新しいハンチントン病の治療戦略を示しました。
東京医科歯科大学・難治疾患研究所/神経病理学分野の岡澤 均教授(脳統合機能研究センター長)の研究グループは、新しい細胞死TRIADの細胞内シグナル経路の詳細を明らかにし、神経変性疾患の一つであるハンチントン病の病態下でTRIADが生じていること、TRIADを標的とすることでハンチントン病の治療が可能であることを示しました。この研究は、カリフォルニア州立大学サンフランシスコ校/グラッドストーン研究所・Steven Finkbeiner教授、レジーナエレーナ国立がん研究所・Giovanni Blandino教授、国立シンガポール大学・Marius Sudol教授、国立精神神経医療研究センター・村田美穂病院長らとの共同研究として行われ、平成26年度から始まった文部科学省『革新的技術による脳機能ネットワークの全容解明プロジェクト』(平成27年度から日本医療研究開発機構:AMEDへ移管)の支援のもとで実施されたもので、その研究成果は、国際科学誌Human Molecular Genetics (ヒューマン モレキュラー ジェネティクス)に、2016年9月13日午前0時(英国時間)にオンライン版で発表されます。
研究の背景と結果の概要
アルツハイマー病、パーキンソン病、ハンチントン病、脊髄小脳失調症、筋萎縮性側索硬化症などの神経変性疾患は、数年から数十年にかけて緩慢に症状が進行することが重要な特徴です。これは、神経細胞の機能低下と細胞死が緩徐に進むためと考えられており、脳梗塞あるいは脳出血など、数分から数時間で症状が完成する脳疾患の細胞死とは明らかに性質を異にするものです。
神経変性疾患の細胞死が如何なるものかは、依然として解明されておらず、議論が続いています。一般に、細胞死には、アポトーシス(注1)、ネクローシスという2つの細胞死があることが有名ですが、近年になって、それ以外に、オートファジー細胞死(注2)、ネクロプトーシス、さらにはTRIAD (Transcriptional Repression-Induced Atypical cell Death, 転写抑制性非典型的細胞死) などの、複数の細胞死が存在することが報告されています。ネクローシスは、本来は熱や強い放射線などの外的要因によって生じる受動的で急速な細胞死を意味しており、細胞質の膨満・破裂、ミトコンドリア膨化などを形態的特徴としていました。Tumor Necrosis Factor(TNF)は、(名前と異なり) がん細胞にアポトーシスを誘導するものとして長らく信じられてきましたが、近年、ネクローシス様の細胞死を誘導し、しかもアポトーシス阻害剤を加えるとネクローシス様細胞死が増加することが報告され(Vercammen et al, J Exp Med 1998)、さらにその細胞内シグナルがRIP1/3というリン酸化酵素(キナーゼ)分子によって伝達されることが明らかになり、ネクロプトーシスと呼ばれるようになりました(Degterev et al, Nat Chem Biol 2005; Degterev et al, Nat Chem Biol 2008, He et al, Cell 2009)。
これに対して、岡澤教授らは、2006年に転写の基本的分子であるRNA polymerase Ⅱの特異的阻害(注3)によって神経細胞に非常に緩慢なネクローシス様細胞死が生じることを発見し、TRIADと命名しました(Hoshino et al, J Cell Biol 2006)。この際に、TRIADはアポトーシスの形態学的・生化学的特徴は持たないこと、オートファジー細胞死とは異なりオートファゴゾームの拡大・増加はなく、小胞体が顕著に膨張していることが形態学的な特徴であること、網羅的発現解析から得た候補分子YAPの関与が疑われることを、併せて報告しました(Hoshino et al, J Cell Biol 2006)。さらに、ショウジョウバエライブラリーを用いてTRIADへ影響する細胞死関連分子をスクリーニングし、得られた分子をタンパク質間相互作用のビッグデータにはめ込むバイオインフォマティクス解析を行うことで、TRIADのシグナルネットワークを包括的に探索し、hnRNPというRNA結合分子、さらにはハンチンチンというハンチントン病原因遺伝子がTRIADに関わっていることを報告しました(Mao et al, Cell Death Dis 2016)。
しかし、TRIADとネクロプトーシスとの関連、TRIADが神経変性疾患に関与するか、などの疑問が残っていました。
結果1.
先行研究において、岡澤教授の研究グループは、DNAからRNAへの転写を司る中核分子であるRNA polymeraseⅡをα-amanitinという特異的阻害剤を用いて抑制し、その際に生じる細胞死TRIADを発見し、疾患との関連性を示唆しました。これに対して、今回の研究では逆方向から調べました。つまり、病気を起こす変異型のハンチンチンを神経細胞に発現させて、どのような細胞死が誘導されるかを調べました。この結果、アポトーシスやネクローシスではなく、不均一な細胞質膨張を特徴とする細胞死が増加していることを見出しました。次に、この細胞質膨張の原因を、各種のlive imaging(注4)を用いて解析し、その本態が小胞体膨張であることを確定しました。また、同様の技術を2光子顕微鏡によるマウス脳での神経細胞の観察に改変し、生きた脳での小胞体の大きさの変動を観察することにも成功しました。変異型ハンチンチン過剰発現マウス(トランスジェニックマウス)と変異型ハンチンチンノックインマウスを対象に解析を行うと、生きたハンチントン病モデルマウスの脳でも、同様に小胞体の不安定化や膨張が観察されました。
さらに、種々の実験を行って、この細胞死を調べたところ、RIP1/3キナーゼの阻害には反応しないが、YAPあるいはそのアイソフォームを発現させることで細胞死を抑制できることが分かりました。このことから、変異型ハンチンチンによって誘導される細胞死は、形態学的にも生化学的にもTRIADと同一のものと結論しました。
結果2.
次に、変異型ハンチンチンによって誘導されるTRIADの細胞内シグナルの詳細を解析しました。YAPはp73、TEADなどの転写因子の活性を制御する転写補助因子として知られています。今回の研究から、分裂細胞で活性の高いPlk1というキナーゼがYAPの77番目のスレオニン(Thr77)をリン酸化することで、YAPの相手をTEADからp73に変換する作用があることが解明されました。つまり、通常Plk1活性が低い神経細胞においては、YAPの相手は主にTEADであり、p73をノックダウンによって減らしてもTRIAD抑制効果は薄いこと、TEADをノックダウンするとTRIADが誘導されること、逆に核内部のYAPの増加させる処理(YAPの過剰発現、Hippoシグナル経路(注5)の抑制など)は、すべて、TRIADを抑制することも分かりました。また、変異ハンチンチンはYAPと結合して封入体に取り込むこと、変異ハンチンチンが発現するとTEADによる転写機能が低下することも分かりました。以上から、TRIADのより詳細なシグナル経路が明らかになりました(図1)。
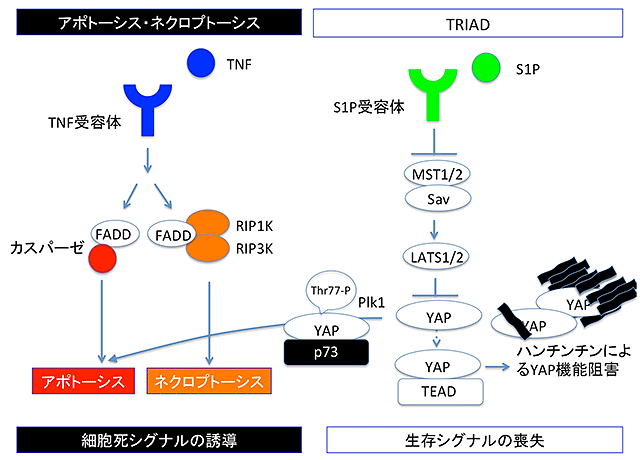
結果3.
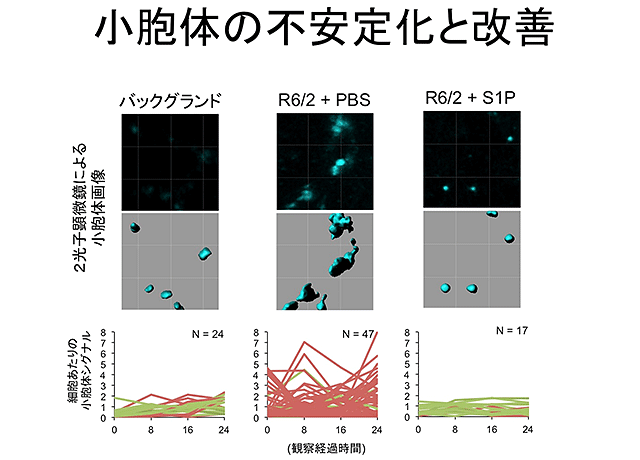
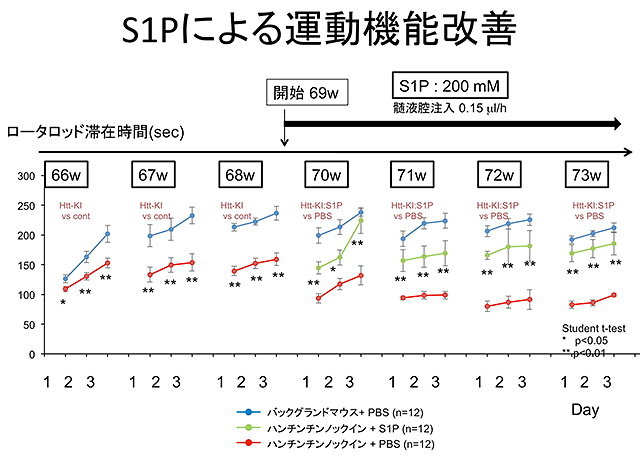
研究成果の意義
用語の解説
- 注1:アポトーシス
- 特定の発生段階で生じる細胞死として見出されたもので、手の水掻きの脱落、脳神経系の過剰な神経細胞の除去などの際に起きる細胞死として有名である。TNFなどの細胞外因子からの受容体を介したシグナル、ミトコンドリアからのcytochrome C放出、小胞体からのシグナルなど複数の経路から始まり、カスパーゼと呼ばれるタンパク質分解酵素が共通して活性化されることによって生じる。DNAが180塩基対前後に断片化されること、核の凝縮・断片化が特徴的で、ミトコンドリアの形態は概ね保たれる。細胞は全体に縮小するかたちで死を迎える。
- 注2:オートファジー細胞死
- 細胞の自己貪食であるオートファジー、特にオートファゴゾーム形成を伴うマクロオートファジーに似た形態を示す細胞死のこと。シグナル経路の詳細はやはりよくわかっていないが、TRIADとは逆にHippo pathway活性化がオートファジー細胞死を抑制するとの報告がある。またアポトーシスに関係するJNKが関与するとの報告がある。
- 注3:RNA polymerase Ⅱ特異的阻害
- RNA polymerase ⅡはゲノムDNAからmRNAを転写する酵素であり、ノーベル賞受賞者のR. Kornberg博士により詳細な構造解析が行われている。RNA polymerase ⅡがDNA2本鎖をまたぐ部分に、α-amanitinがはまり込むこともKornberg博士が示している。
- 注4:live imaging
- 生きた細胞のダイナミックな変化を光学顕微鏡などで連続的に撮影すること。蛍光融合タンパク質などを利用し特定のタンパク質の細胞内動態や酵素の活性化を検出することも可能である。
- 注5:Hippoシグナル経路
- 体の大きさに異常のあるショウジョウバエ変異体から取られた遺伝子Hippo(哺乳類Mst)を皮切りに、その上下のシグナル分子が解明されて明らかになった細胞内シグナル経路のこと。Hippシグナルの活性化とは、経路中の複数のキナーゼが活性化され、YAPあるいはTAZのリン酸化を起こす。この結果、YAPは14-3-3タンパク質にトラップされて核移行が阻害される。
- 注6:
- S1P:スフィンゴシン−1—リン酸、sphingosine-1-phosphate。細胞膜を構成するリン脂質であり、Gタンパク質共役受容体であるS1P受容体と結合して各種の細胞反応を引き起こす。多くの場合、細胞増殖や生存シグナルにつながると考えられる。
問い合わせ先
研究に関すること
東京医科歯科大学 難治疾患研究所・脳統合機能研究センター
神経病理学分野 岡澤 均(オカザワ ヒトシ)
TEL:03-5803-5847 FAX:03-5803-5847
E-mail:okazawa.npat“AT”mri.tmd.ac.jp
AMED事業に関すること
日本医療研究開発機構 脳と心の研究課
〒100-0004 東京都千代田区大手町1-7-1
TEL:03-6870-2222 FAX:03-6870-2244
E-mail:brain“AT”amed.go.jp
報道に関すること
東京医科歯科大学 広報部広報課
〒113-8510 東京都文京区湯島1-5-45
TEL:03-5803-5833 FAX:03-5803-0272
E-mail:kouhou.adm“AT”tmd.ac.jp
※E-mailは上記アドレス“AT”の部分を@に変えてください。
掲載日 平成28年9月13日
最終更新日 平成28年9月13日