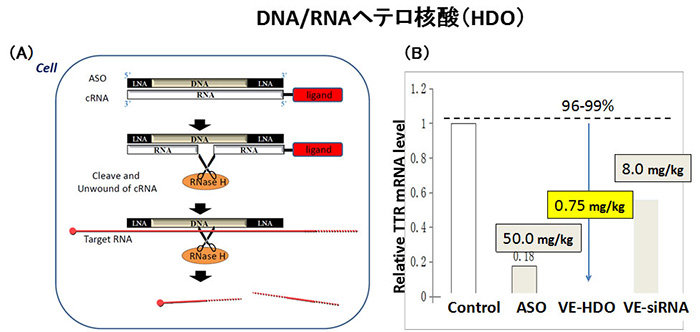
<図1> DNA/RNAヘテロ核酸(HDO)の遺伝子抑制メカニズム(A)と有効性(B)
(A)Ligand分子で細胞内へ導入されたヘテロ核酸は、RNaseHによって相補鎖RNA(cRNA)が切断され単独のアンチセンス核酸(ASO)となる。その後、標的mRNAに結合して、遺伝子発現抑制効果を生じる。
(B)その有効性はASOやsiRNAよりはるかに高い。
ホーム > 平成26年度採択課題 > 第3世代ヘテロ核酸の開発
研究開発代表者:横田 隆徳
東京医科歯科大学 大学院医歯学総合研究科 教授

核酸医薬品は標的遺伝子への特異性が高いことから次世代バイオ医薬品として期待されていますが、核酸医薬の主流であるアンチセンス核酸(ASO)、siRNAは基本特許が欧米に抑えられていて、日本は大きく遅れをとっており、現状の技術では世界をリードする創薬は困難です。 日本での核酸医薬開発の課題(ボトルネック)の解決には、1)細胞内の標的をより効率的に制御する日本独自の基盤技術、2)肝臓以外の特定の臓器および細胞への送達する技術、3)経口や経皮など侵襲度の低い投与を可能とする技術開発が必要と考えられます。
我々は最近、ASOやsiRNAとその分子構造、作用機構が異なる第3の核酸医薬であるDNA/RNA2本鎖ヘテロ核酸を創生しました。 2本鎖ヘテロ核酸は標的mRNAに結合するアンチセンスgapmer (LNA-DNA-LNA) の主鎖と、主鎖に相補的なRNA (cRNA)からなる非天然機能核酸です。この主鎖は両端がLNA、中央部がDNAで、2本鎖の中央部がDNA-RNAヘテロ核酸になるため、この部分が細胞内のエンドヌクレアーゼであるRNase Hによって相補鎖RNAが切断されます。 その結果、単独となった主鎖が標的mRNAに結合して再びRNase Hが標的mRNAを切断して遺伝子抑制効果を発揮するデザインです。 すなわち、RNase Hが相補鎖RNAと標的mRNAの切断の一人二役を果たすことにより、主鎖の結合親和性に影響を与えることなく相補鎖RNAに誘導分子を結合することが可能となった点が特徴の分子技術です(図1A)。
2本鎖ヘテロ核酸の特徴である内在型の薬剤送達システムの導入分子として、我々の特許であるビタミンE (VE)をリガンド分子として結合させることにより、静脈投与で従来のASOや、VE結合siRNA(VE-siRNA)より標的遺伝子抑制効果の飛躍的な上昇に成功し、 さらにその抑制率も低投与量(0.75mg/kg)でも99%以上と今までにない劇的な抑制率を達成しました。その有効性は現状の核酸医薬で最高水準です(図1B)。
DNA/RNA2本鎖ヘテロ核酸はASOやsiRNAとその分子構造・作用機構が異なって、その有効性は臓器へのデリバリー効果のみでなく、細胞内での特有のプロセシング経路による増強効果があることが想定されており、 その背景には生体内での生物学的に新規の機序があると考え、第3の核酸医薬であると考えています。
さらに異なる分子設計によってmRNA以外にもマイクロRNAなどの非コードRNAやRNA編集制御も可能とする第2世代も引き続いて開発しました。第2世代は従来のマイクロRNA阻害薬の10倍以上の有効性をしめしました。
しかし、第1世代/第2世代ヘテロ核酸においては、その効果は肝臓に限定されており、その投与ルートも静脈投与のみで有効でした。 最近、我々その新規の分子設計と投与方法によって肝臓以外の心筋、腎臓などほとんどの多くの腹部臓器や皮下脂肪、骨格筋の標的遺伝子制御が可能になりました(図2)。 しかし、肝障害の副作用があり実用には不十分です。本研究では、東京大学の宮田完二郎博士と共同して肝臓へのnegative targetingと標的臓器へのpositive targetingによって副作用なく目的臓器の遺伝子制御を可能とする新規の基盤技術である第3世代ヘテロ核酸の創生を目的とします。
さらにヘテロ核酸の臨床応用性を拡大させるために、非侵襲的投与ルートの開発を合わせて試みたいと考えています。 腸管、皮膚上皮には上皮細胞間をシールするタイトジャンクションが存在して、高分子薬の侵入をブロックする上皮バリアを形成しています。 大阪大学の八木清仁/近藤昌夫博士の開発したタイトジャンクション制御分子を応用し、最適化して経口、経皮投与方法の開発を合わせて行います。

(A)Ligand分子で細胞内へ導入されたヘテロ核酸は、RNaseHによって相補鎖RNA(cRNA)が切断され単独のアンチセンス核酸(ASO)となる。その後、標的mRNAに結合して、遺伝子発現抑制効果を生じる。
(B)その有効性はASOやsiRNAよりはるかに高い。
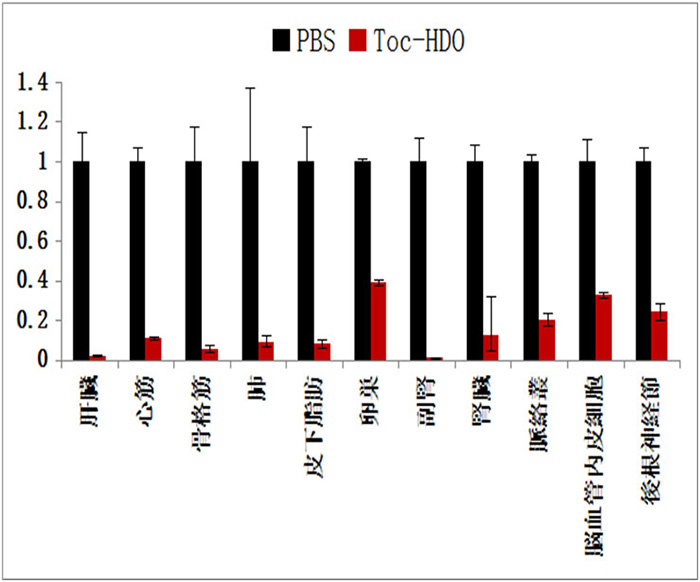
ヘテロ核酸静脈投与により肝臓以外の多くの腹部臓器で内因性標的遺伝子制御が可能になった。