

B-cell signaling factors essential for immunological memory in infections and vaccines discovered
News Releases & Research Results
The University of Tokyo
Osaka University
Japan Agency for Medical Research and Development
Research Background
Current COVID-19 pandemic has brought into our attention that there are several unknown evasion mechanisms by pathogens. To protect host from infection/re-infection, immunological memory must be elevated via one time infection or vaccination. Although this is the main study area of immunology field, however, little is known how immune memory is developed and how long it is durable during infection and vaccination. As an example, malaria infection is one of the most troublesome infections in the world and known that an individual remains susceptible to repeated malaria infection. To date, how malaria infection impairs immune memory that confers sterile protection is not fully understood.
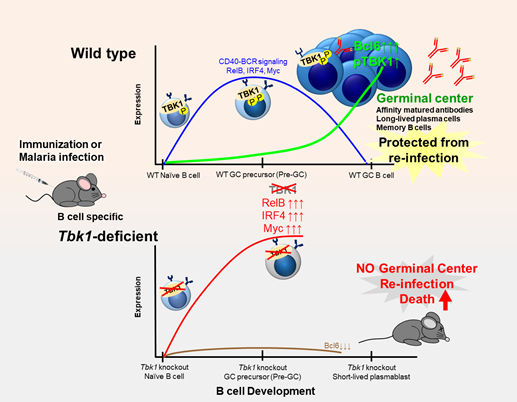
Germinal centers (GC) are highly dynamic structures formed in lymphoid organs and are sites where somatic hypermutation and clonal selection are coupled for high-quality durable antibody responses leading immune memory. The anatomical structure and function of GCs have been studied for a long, but only recently information has been gathered about how B cells are guided through the development of GCs. Upon antigen encounter, B cells rapidly proliferate and differentiate into multipotent precursor cells that have the potential to diverge into two distinct fates: either GC or extrafollicular (EF) responses. Induction of GC reaction is often the desirable outcome of an infection or vaccine to provide antigen-specific long-lasting humoral immunity. Conversely, the induction of GC-independent EF responses in several disease models and vaccinations results in early acute production of short-lived plasmablasts. The intrinsic and extrinsic factors that determine the fate of antigen-activated B cells, whether to enter GC reaction or to commit to EF responses are not yet well-defined.
TANK-binding kinase 1 (TBK1) is a serine/threonine kinase that is famously known for its role in innate immunity especially for type I interferon (IFN-I) production upon nucleic acid recognition. Apart from its role for IFN-I production via phosphorylation of IRF3/7, TBK1 is also involved in various signaling pathways in a cell-type specific manner. Earlier studies found that TBK1 is among one of the genes upregulated in GC B cells upon NP-CGG immunization, suggesting that TBK1 may have a role in GC formation. However, whether TBK1 plays a critical role in GC-dependent memory responses have remained un-answered.
Outline of research results
Here, using both malaria infection and immunization models, we identified TBK1 as a novel B cell intrinsic factor that is crucial in regulating GC differentiation. We found that phosphorylation of TBK1 increased in B cells during GC differentiation (Figure). Notably, TBK1 deletion in B cells led to impaired GC formation and therefore increased susceptibility to a non-lethal malaria infection. Memory B cells generated in the absence of TBK1-dependent GC failed to confer sterile immunity upon re-infection challenge. Although TBK1 deficient B cells could be activated to differentiate into GC precursor cells (Pre-GC), the fate of these cells was distinct from that of TBK1 sufficient B cells. We found that TBK1 negatively regulates both CD40 and BCR signaling in Pre-GC. Mechanistically, increased CD40-induced RelB expression in Tbk1-deficient Pre-GC enhanced IRF4 expression that subsequently suppressed Bcl6 expression, the master regulator of GC formation. Unlike naïve B cells, BCR activation further enhanced CD40-induced IRF4 expression in Pre-GC in a TBK1-dependent manner. This study revealed a crucial role of B cell intrinsic TBK1 as a determinant of GC commitment through the fine tuning of CD40 and BCR signaling (Figure).
Future prospects
Overall, our findings are expected to have crucial implications for tailoring successful vaccines to produce long lasting protective immunity against several problematic infections like malaria as well as a countermeasure to future pandemics.
TBK1 is required for IFN-I mediated antiviral immunity, while here we additionally showed its importance in GC-mediated humoral immunity. However, TBK1 is also involved in the pathogenesis of autoimmune diseases. Moreover, TBK1 inhibitor, such as Amlexanox has been used in clinical treatment of asthma and recently been proposed for the treatment of obesity related metabolic diseases. Our findings suggest that TBK1 inhibitors should be administered with caution, considering its possible effect on humoral immunity in case of infection and vaccination. Moreover, the timing of TBK1 inhibitor administration may be important to target TBK1 in specific cell types during disease progression and to prevent its suppressive effects on vaccine efficacy.
Publication details
- Title
- B cell intrinsic TBK1 is essential for germinal center formation during infection and vaccination in mice
- Journal
- Journal of Experimental Medicine
- Authors
- Michelle S. J. Lee1,12, Takeshi Inoue2, Wataru Ise2, Julia Matsuo-Dapaah1, James B. Wing3,4, Burcu Temizoz5, Kouji Kobiyama5, Tomoya Hayashi5, Ashwini Patil6, Shimon Sakaguchi7, A. Katharina Simon8, Jelena S. Bezbradica8, Satoru Nagatoishi9, Kouhei Tsumoto9,10, Jun-Ichiro Inoue9, Shizuo Akira11, Tomohiro Kurosaki2, Ken J. Ishii5,11,12, Cevayir Coban1,11,12*
-
- Division of Malaria Immunology, Department of Microbiology and Immunology, The Institute of Medical Science (IMSUT), The University of Tokyo, Tokyo, Japan
- Laboratory of Lymphocyte Differentiation, Immunology Frontier Research Center (IFReC), Osaka University, Osaka, Japan
- Laboratory of Human Immunology (Single Cell Immunology), IFReC, Osaka University, Osaka, Japan
- Human Single Cell Immunology Team, Center for infectious Disease Education and Research (CiDER), Osaka University, Osaka, Japan
- Division of Vaccine Science, Department of Microbiology and Immunology, The Institute of Medical Science (IMSUT), The University of Tokyo, Tokyo, Japan
- Combinatics Inc., Tokyo, Japan
- Laboratory of Experimental Immunology, IFReC, Osaka University, Osaka, Japan
- The Kennedy Institute of Rheumatology, NDORMS, University of Oxford, Oxford, UK
- Research Platform Office, The Institute of Medical Science (IMSUT), The University of Tokyo, Tokyo, Japan
- Department of Bioengineering, Graduate School of Engineering, The University of Tokyo, Tokyo, Japan.
- IFReC, Osaka University, Osaka, Japan
- International Vaccine Design Center (VDesC), The Institute of Medical Science (IMSUT), The University of Tokyo, Tokyo, Japan
- *Correspondence: [email protected] (C.C.)
- DOI
- 10.1084/jem.20211336
Abstract of the paper
Germinal center (GC) is a site where somatic hypermutation and clonal selection are coupled for antibody affinity maturation against infections. However, how GCs are formed and regulated is incompletely understood. Here, we identified an unexpected role of Tank-binding kinase-1 (TBK1) as a crucial B cell intrinsic factor for GC formation. Using both immunization and malaria infection models, we show that TBK1-deficient B cells failed to enter GC reaction despite the induction of normal TFH differentiation. Mechanistically, TBK1 is increasingly phosphorylated in B cells during GC differentiation and fine tunes the balance between IRF4 and BCL6 expression by limiting CD40 and BCR activation through non-canonical NF-κB and AKTT308 signaling. In the absence of B cell intrinsic TBK1, CD40 and BCR signaling uncontrollably upregulated IRF4 expression in Pre-GC leading to the suppression of BCL6, resulting in the failure to form GC to mount high affinity, and antigen-specific antibodies. As a result, memory B cells generated in the absence of TBK1 fail to confer sterile immunity upon re-infection. These findings have crucial implications for tailoring vaccines to produce long lasting protective humoral immunity.
Contact
Cevayir Coban, Professor, Division of Malaria Immunology, The Institute of Medical Science (IMSUT), The University of Tokyo
UTokyo PEOPLE COBAN Cevayir
Division of Malaria Immunology
AMED Research Program inquiries
Japan Agency for Medical Research and Development (AMED)
Division of Basic Medical Research, Department of Basic Medical Research
20F Yomiuri Shimbun Building, 1-7-1 Otemachi, Chiyoda-ku, Tokyo 100-0004, Japan
E-mail: jprogram"AT"amed.go.jp
Please change the above address "AT" to @
Links
12/16/21
Last updated 12/16/21