

Proposing a new concept that integrates species evolution and genetic evolution
News Releases & Research Results
Chiba Cancer Center
Japan Agency for Medical Research and Development
A research group at the Chiba Cancer Center has proposed a new concept that integrates species evolution and gene evolution which has now been published in EMBO Reports, a scientific journal of the European Molecular Biology Organization. In recent years, human genes that have emerged since the evolutionary divergence from apes have been shown to be involved in the onset and progression of cancer. However, the mechanism by which these evolutionarily novel genes emerged has remained a mystery.
The research group, in collaboration with the National Cancer Center Research Institute and Tohoku University, devised an index for detecting the birth of genes, called ORF dominance, and calculated ORF dominance for 100 organisms selected from all three domains of life (bacteria, archaea, and eukaryotes) to determine how genes have been born during evolution. New human gene candidates identified using this index are suggested to be related to brain development and cancer. We also found that the probability of new gene birth is particularly high in species with small populations, such as endangered species.
The process of cancer development is called "cancer evolution" because DNA mutations create genes that change the composition of the cancer cell population. In the future, we will calculate ORF dominance in cancers to develop a method for suppressing cancer evolution.
Study Outline
A joint research team led by Yusuke Suenaga, Senior Research Scientist at Chiba Cancer Center Research Institute, Mamoru Kato, Director at the National Cancer Center Research Institute, and Professor Takashi Makino of Tohoku University Graduate School of Medicine have devised a novel and useful index to detect newly emerged genes during evolution, called ORF dominance. The research team has elucidated one aspect of the mechanism underlying gene birth by calculating the total RNA of 100 organisms selected from among bacteria, archaea, and eukaryotes.
In recent years, evolutionarily novel human genes that have emerged since the evolutionary diversion from apes have been reported to be involved in various diseases, including cancer, and are attracting attention as potential new therapeutic targets. On the other hand, there are many noise-like RNAs in cells that have no function, and it has traditionally been difficult to efficiently discover RNA of new genes emerging in evolution because they resemble such noise-like RNA.
The index we developed, ORF dominance, was found to be associated with the efficiency with which RNA is translated into protein and is useful for distinguishing noise-like RNA from RNA of newly emerging genes. The new human gene candidates we identified using ORF dominance are suggested to likely be associated with genetic diseases such as holoprosencephaly and glioblastoma, a type of brain tumor. We also show that new genes are more likely to be generated from non-coding RNAs in eukaryotes than in prokaryotes. Furthermore, the probability of gene birth was found to be particularly high in species with small populations, such as endangered species. Based on these results, we proposed a model in which organisms create new genes as a countermeasure against population decline. If population decline is too rapid, the birth of new genes will not be able to contribute to adaptation to the environment, and the species will be driven to extinction.
Key research findings
- We devised a useful index, ORF dominance, to identify evolutionarily novel genes.
- Candidates of newly emerging human gene may be associated with genetic diseases and brain cancer.
- The probability of gene birth is high in species with small populations, especially in endangered species.
Background of the study
According to the central dogma of biology, a protein exerts its function when messenger RNA (mRNA) is transcribed from the DNA region of a gene and translated into a protein (Fig. 1A, red box). Recently, however, RNAs that can function without being translated into proteins have been discovered; these are called non-coding RNAs (Fig. 1A, right). These non-coding RNAs include ribosomal RNAs and transfer RNAs, which are known conventionally, as well as RNAs with various functions such as transcriptional regulation and maintenance of the nuclear structure. On the other hand, the functions of most of the non-coding RNAs have not been discovered, suggesting that they may be essentially functionless, acting like noise (Fig. 1A, right). In contrast to non-coding RNAs, mRNAs that are translated into proteins are also called coding RNAs (Fig. 1A). In other words, genes are broadly understood to be DNA regions that transcribe coding RNAs or non-coding RNAs with functions (Fig. 1A).
However, in known coding RNAs, such as the RNA of the tumor-suppressor gene p53, it has been demonstrated that the RNA per se has physiological functions. Conversely, there are many reports that RNAs usually known as non-coding RNAs have been translated into proteins, and the physiological functions of the proteins have been demonstrated.
Such RNAs cannot be categorized based on the binary distinction between coding and non-coding RNAs, and are called bifunctional RNAs (Fig. 1B). The existence of bifunctional RNA suggests that the boundary between coding/non-coding RNAs is inherently ambiguous and rather continuous. However, it is unclear what properties of RNAs are responsible for this ‘continuity.’
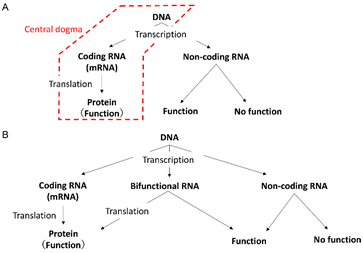
- The relationship between the central dogma and non-coding RNA. In the central dogma (red box), genes are regions of DNA that transcribe coding RNAs, but in a broader sense, genes also include regions of DNA that transcribe non-coding RNAs with functions.
- The existence of bifunctional RNA challenges the dualistic distinction between coding and non-coding RNA.
Yusuke Suenaga, a senior researcher at Chiba Cancer Center Research Institute, and his colleagues previously discovered the existence of a gene called NCYM in the complementary strand of oncogene MYCN in the process of studying neuroblastoma, a pediatric tumor (Suenaga et al., PLoS Genetics 2014). New genes have previously been thought to arise from pre-existing genes (Fig. 2A), but NCYM is thought to have evolved from a non-genic region that transcribes non-coding RNA into a gene that transcribes coding RNA (Fig. 2B). Such genes are called de novo genes, and NCYM was the first human de novo gene to demonstrate physiological functions under experimental conditions. Furthermore, NCYM was reported to function as a non-coding RNA, indicating that it is a bifunctional RNA (Suenaga et al., Jpn J Clin Oncol 2020).
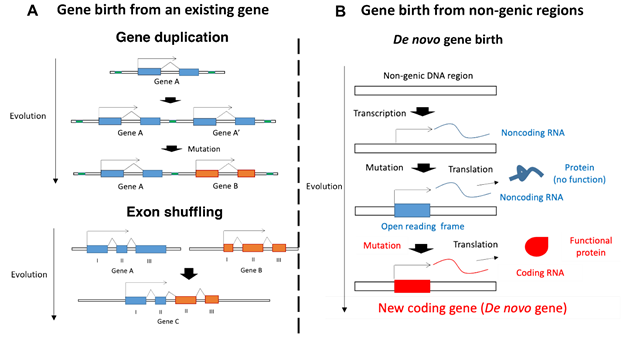
- Gene birth from an existing gene. In the upper panel, gene A is duplicated to give rise to gene A', which accumulates mutations to become the new gene B (gene duplication). The lower panel shows the process of formation of gene C from some exons of genes A and B (exon shuffling).
- Gene birth from non-genic regions. Noise-like non-coding RNAs transcribed from non-genic regions become non-coding RNAs that are translated into non-functional proteins by acquiring a reading frame through the accumulation of mutations, and then coding RNAs that are translated into functional proteins are born through further mutations. Genes created in this way are called de novo genes.
Since NCYM is an emerging gene in evolution, the research group hypothesized that examining the RNA of NCYM could help explain the ‘continuity’ in the distinction between non-coding and coding RNAs.
Research Contents
We focused on the RNA sequence to explain this ‘continuity.’ Coding RNAs and bifunctional RNAs are translated into proteins by ribosomes in the cell, and when we compared the RNA sequence of NCYM with those of non-coding RNAs, we noticed that there were few extra ORFs besides the reading frame (ORF), the sequence through which ribosomes translate proteins.
Therefore, we defined ORF dominance as an index that mathematically expresses the low number of extra open reading frames (ORFs) (Fig. 3).
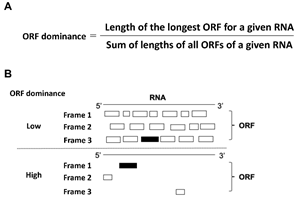
- Formula defining ORF dominance.
- ORF status of RNAs with low and high ORF dominance. Black rectangles represent the longest ORFs; white rectangles are superfluous ORFs. Ribosomes translate RNA into proteins in three frames in the 5' to 3' direction.
This ORF dominance can be calculated for any RNA sequence. The ORF dominance of coding RNAs (red in Fig. 4) and non-coding RNAs (blue in Fig. 4) exhibited high values and low values, respectively, across all three domains (bacteria, archaea, and eukaryotes; Fig. 4). In bacteria and archaea, the boundary between coding and non-coding RNAs was clear, whereas in eukaryotes, there were many RNAs that showed intermediate ORF dominance values and the boundary between coding and non-coding RNAs became ambiguous. This suggests that new bifunctional RNAs and coding RNAs are more likely to be generated from non-coding RNAs in eukaryotes. The ORF dominance of the tumor-suppressor gene p53 showed intermediate values, reflecting its bifunctional nature.
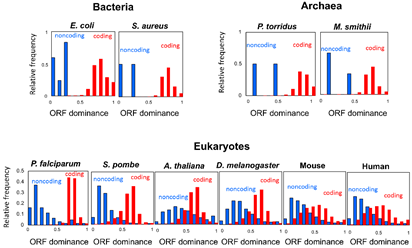
Human non-coding RNAs with high ORF dominance are candidates for de novo genes and have been associated with transcription factors such as MYCN, TGIF, and ZIC2, and with glioblastoma, a type of brain tumor. MYCN is an oncogene involved in neuroblastoma and also causes Feingold syndrome, a genetic disease that causes brain atrophy. TGIF and ZIC2 are also causative genes of holoprosencephaly, a genetic disorder in which the higher functions of the brain are impaired. These results suggest that human de novo gene candidates are associated with brain development and tumorigenesis.
We further defined Odom, a value which mathematically represents the ambiguity of the boundary between coding and non-coding RNAs. This boundary ambiguity, Odom, is calculated from the distribution of ORF dominance, with one value for each species. Odom was positively and negatively correlated with the genomic DNA mutation rate and effective population size (number of individuals) of the species, respectively. Odom was found to be higher for Red List threatened species, and was higher for non-threatened species with declining populations than for species with stable populations. These results suggest that DNA mutation rates increase as populations decline, blurring the coding/non-coding RNA boundary and increasing the probability of gene birth. In other words, organisms may be creating genes as a countermeasure against population decline (Fig. 5).
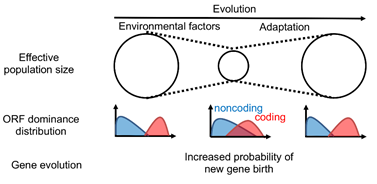
On the other hand, an intermediate ORF dominance implies the loss of characteristics of coding and non-coding RNA sequences, and a higher risk of becoming a non-functional non-coding RNA. In other words, the probability of gene loss also increases. Rapid population decline may cause lethal gene loss before new gene birth contributes to environmental adaptation, which may lead to the extinction of the species.
In summary, ORF dominance is a useful index for understanding species evolution and gene evolution in a unified manner.
Significance and application of this study
Cancer cells accumulate mutations in their DNA during their development and progression, giving rise to oncogenes. ORF dominance may be useful in predicting which fusion gene RNAs are being translated and driving cancer progression. In addition, proteins translated from non-coding RNAs are presented on the cell surface as neoantigens in cancer cells, and are targets for tumor immunity. If ORF dominance proves to be useful in the identification of neoantigens, it will contribute to predicting the efficacy of immune checkpoint inhibitors and determining the expected efficacy of cancer immunotherapy. The blurring of the coding/non-coding RNA boundary, Odom, is related to the number of individuals of a species, which may be useful for predicting extinction in evolutionary and conservation biology. For example, even if it is difficult to estimate the population size of a rare species through ecological studies, ORF dominance can be calculated by examining the total RNA sequences of a small number of individuals, and may assist with estimating population size and risk of extinction.
Collaborative Research Team
Mamoru Kato, Director, Department of Bioinformatics, National Cancer Center Research Institute
Professor Takashi Makino, Department of Evolutionary Genomics, Graduate School of Life Sciences, Tohoku University
Research Support
This study was supported by:
The Japan Agency for Medical Research and Development (AMED)
International Joint Research and Development Promotion Project on Science and Technology in Medical Fields Interstellar Initiative (18jm0610006h0001)
The Yasuda Memorial Foundation for Medical Science Grant for Young Cancer Researchers
Takeda Foundation for Advancement of Science, Grant-in-Aid for Medical Research
Original Research Paper Information
- Title
- Open reading frame dominance indicates protein-coding potential of RNAs
- Authors
- Yusuke Suenaga1,*,† , Mamoru Kato2,† , Momoko Nagai2 , Kazuma Nakatani1,3,4 , Hiroyuki Kogashi1,3 , Miho Kobatake1 , Takashi Makino5
- Author affiliations
-
- Department of Molecular Carcinogenesis, Chiba Cancer Centre Research Institute, Chiba, Japan
- Division of Bioinformatics, National Cancer Centre Research Institute, Tokyo, Japan
- Department of Molecular Biology and Oncology, Chiba University School of Medicine, Chiba, Japan
- Innovative Medicine CHIBA Doctoral WISE Program, Chiba University School of Medicine, Chiba, Japan
- Laboratory of Evolutionary Genomics, Graduate School of Life Sciences, Tohoku University, Sendai, Japan
- *. Responsible author
- †. Equal contribution
- Journal
- EMBO Reports, the scientific journal of the European Molecular Biology Organization
- DOI
- 10.15252/embr.202154321
- Date of publication
- Tuesday, April 19, 2022 (EMBO Reports)
- URL of the article
- https://www.embopress.org/doi/10.15252/embr.202154321
Contact
For inquiries, please contact:
In relation to this study
Department of Carcinogenesis Control Research, Chiba Cancer Center Research Institute
Yusuke Suenaga, Senior Researcher
E-mail: ysuenaga"AT"chiba-cc.jp
Telephone: +81-43-264-5431
About the press release
Medical Management Division, Chiba Cancer Center
Yusuke Ono
E-mail: y.on34"AT"pref.chiba.lg.jp
Telephone: +81-43-264-5431
Matters related to AMED's projects
Interstellar Initiative for the Promotion of International Scientific and Technological Cooperative Research and Development in Medical Fields
Office of International Collaboration, International Strategy Promotion Division, International Strategy Promotion Department
E-mail: interstellar"AT"amed.go.jp
Telephone: +81-3-6870-2216
Replace “AT” with “@”
04/20/22
Last updated 04/20/22