


プリオノイドを促進するRNAの形「G4」―新たな神経変性治療ターゲットの発見―
プレスリリース
熊本大学
日本医療研究開発機構
ポイント
- 遺伝性の神経疾患「脆弱X随伴振戦/失調症候群(FXTAS)」の発症には、原因遺伝子の異常な繰り返し配列が形成するRNAの特殊な構造「グアニン四重鎖構造(G4)」が関与することを明らかにした。
- G4はプリオノイドタンパク質「FMRpolyG」の凝集を促進し、神経障害を引き起こすことを発見し、この病態発症メカニズムを「G4プリオノイド」とした。本メカニズムに基づき、「5-アミノレブリン酸」が神経障害(神経変性)の治療に有効であることが示唆された。
- 「G4プリオノイド」はFXTASだけでなく、アルツハイマー病やパーキンソン病に共通するメカニズムであることが予想され、5-アミノレブリン酸について今後治療薬の適応拡大が期待される。
概要説明
熊本大学発生医学研究所の塩田倫史独立准教授、矢吹悌助教、朝光世煌研究員らの研究グループは、「脆弱X随伴振戦/失調症候群(FXTAS)」※1における神経機能異常のメカニズムおよび治療候補薬を発見しました。
FXTASは、国の難病に指定されている遺伝性の神経変性疾患で、原因遺伝子の一部にある繰り返し配列(CGGリピート)数の異常によって引き起こされます。塩田准教授らは、解析の結果、CGGリピートRNA由来の「グアニン四重鎖構造(G4)」※2が、プリオノイドタンパク質※3のひとつである「FMRpolyG」の凝集・細胞間伝播の核となり、神経機能の異常を引き起こすことを明らかにし、この病態発症メカニズムを「G4プリオノイド」としました。
また、「プロトポルフィリンIX」という物質が、G4に結合してFMRpolyGの凝集を抑制するとともに、FMRpolyGの産生も抑制することを明らかにしました。そこで、体内のポルフィリン合成経路によってプロトポルフィリンIXに代謝される薬剤「5-アミノレブリン酸」をFXTASモデルマウスに経口投与し、モデルマウスで見られる神経機能異常が改善されるか検討しました。結果として、5-アミノレブリン酸はFXTASモデルマウスで低下した神経伝達機能・認知機能・運動機能を有意に改善することが分かりました。5-アミノレブリン酸は安全性の高い既承認医薬品であり、神経難病である「ATR-X症候群」における認知機能の低下にも有効であることが既存の研究により示されています(参考文献1、2)。このことから、FXTASの神経機能異常に対しても改善効果を示すことが期待されます。
なお、近年、ゲノム解析によりFXTASと同様のCGGリピートに由来する疾患(トリプレットリピート病)が多数報告されていますが、本研究により発見した病態発症メカニズムを基盤として、トリプレットリピート病に対する治療の進展が期待されます。さらに、アルツハイマー病やパーキンソン病等の神経変性疾患には、プリオノイドが関与することが示唆されています。従って、本研究により発見されたG4によるプリオノイド機構「G4プリオノイド」を基盤とした神経変性疾患に対する治療が期待されます。
本研究成果は、日本医療研究開発機構(AMED)難治性疾患実用化研究事業(JP19ek0109235;JP20ek0109425)、MEXT/JSPS科研費(JP16K08265;JP19H05221;JP20H03393;JP20K15417;JP20J00520;JP19K16369)、アステラス病態代謝研究会、第一三共生命科学研究振興財団、かなえ医薬振興財団、三菱財団、国立大学法人熊本大学国際先端研究拠点およびトランスオミクス医学研究拠点ネットワーク形成事業の支援を受けて、米国科学振興協会(AAAS)が発行する科学誌「Science Advances」に令和3年1月13日午後2時(米国東部標準時)(日本時間令和3年1月14日午前4時)にオンライン掲載されます。
背景・内容
脆弱X随伴振戦/失調症候群(Fragile X-associated tremor/ataxia syndrome;FXTAS)は、国の難病に指定されている遺伝性の神経変性疾患で、原因となる遺伝子に含まれる繰り返し配列(CGGリピート)数が異常に長くなってしまう(正常は45以下であるリピートが、55~200リピートに拡張する)ことによって引き起こされます。FXTASの症状として、手足のふるえや身体のふらつきといったパーキンソン症候群の症状、および認知機能の低下が見られ、多くは50歳以降に発症し、有病率は年齢とともに増加します。米国の調査では、男性の403人に1人、女性の209人に1人が55~200のCGGリピートを持っており、そのうち40%の男性、16%の女性がFXTASと診断されています。
FXTASの発症要因は長年不明でしたが、最近の研究により、2つの主要な病態メカニズムが提唱されました(参考文献3)。1つは「RNA毒性」による神経障害で、異常な長さのCGGリピート部分のRNAに様々なRNA結合タンパク質が集積し、神経機能の障害を引き起こすというメカニズムです。もう一つは、「RAN(Repeat-Associated Non-AUG)翻訳」※4による神経障害で、異常な長さのCGGリピートを持つ遺伝子に基づいて作られた病原性のタンパク質FMRpolyGが神経機能障害を引き起こすというメカニズムです(図1)。
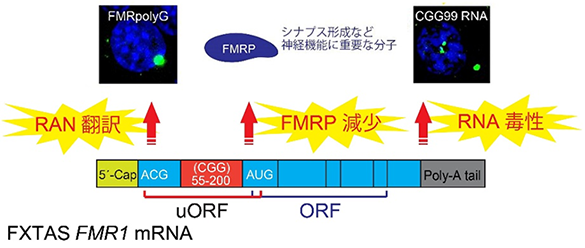
しかしながら、FXTASにおける神経障害の主要な発症要因として、上記のどちらが、もしくは両方が重要なのか、大きな論争になっていました。今回、本研究グループはこの問題を解決し、新たな病態発症メカニズムである「G4プリオノイド」を提唱しました。
成果
1)FMRpolyGはCGGリピートRNA由来のG4により凝集する
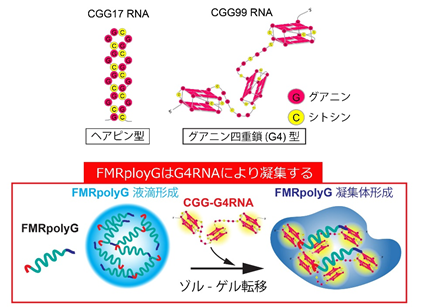
本研究では、RAN翻訳により産生されるタンパク質FMRpolyGに含まれるグリシン連続領域にプリオン様の性質があることに着目しました。プリオン様領域を持つタンパク質は液-液相分離(LLPS)※5により液滴を形成しますが、FMRpolyGの液滴は、CGG99リピート(FXTAS疾患発症リピート数)RNAと結合し、ゲル状の凝集体を形成しました。
そこで、RNAの物性解析を行ったところ、健常人のリピート数であるCGG17リピートRNAがヘアピン型であるのに対し、CGG99リピートRNAはG4を形成しており、FMRpolyGのグリシン連続領域と直接結合しました。つまり、FMRpolyGはグリシン連続領域を介して、G4RNAと結合することで凝集することが分かりました(図2)。
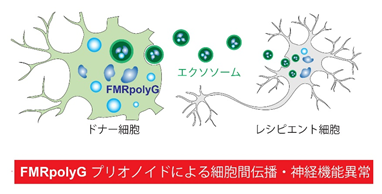
2)FMRpolyGはエクソソーム(細胞外小胞)を介して細胞間伝播し、神経機能異常を引き起こす
次に、FMRpolyGに結合するタンパク質を網羅的に解析しました。その結果、FMRpolyGは、神経変性疾患の発症に関与することが示されているRNA結合タンパク質群(HNRNPA2/B1、FUS、SFPQ等)と結合することが分かりました。また、FMRpolyGは細胞外小胞であるエクソソームに含まれるタンパク質群とも複合体を形成することが分かりました。実際に、FXTASモデルマウスの脳由来エクソソームにはFMRpolyGが高発現しており、野生型マウス神経細胞(レシピエント細胞)にFXTASモデルマウス神経細胞(ドナー細胞)由来エクソソームを処置することによって、野生型マウス神経細胞にもFMRpolyGの発現が確認され、神経機能異常が見られました。つまり、FMRpolyGはエクソソームを介して細胞間に伝播し、神経機能異常を引き起こすプリオノイドタンパク質であることが分かりました(図3)。
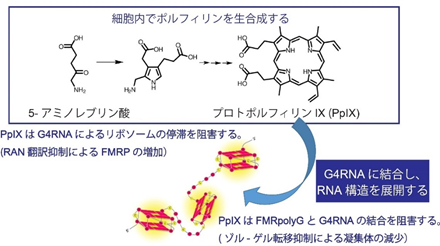
3)G4作用薬「5-アミノレブリン酸」はFMRpolyGのRAN翻訳と凝集を抑制し、FXTASモデルマウスにおける神経障害を改善する
さらに、これまで本研究グループは、「5-アミノレブリン酸」により、細胞内でG4に結合する性質を持つポルフィリン類がつくられることを見出しており、そのひとつである「プロトポルフィリンIX」をFMRpolyGとG4RNAが結合した凝集体に処置することで、凝集体が劇的に縮小することを発見しました(図4)。これは、G4RNAにプロトポルフィリンIXが結合し、FMRpolyGとG4RNAの結合を阻害したためと考えられます。
さらに、5-アミノレブリン酸をFXTASモデルマウスに経口投与したところ、FMRpolyGの発現が抑制され、代わりに神経機能に重要なタンパク質(FMRP)の発現が増加することで、低下した神経可塑性・認知機能・運動機能を有意に改善できることを明らかにしました。
展開
本研究では、FXTASにおけるCGGリピート由来RNAがG4を形成しFMRpolyGに結合することで、FMRpolyGの細胞間伝播「プリオノイド」を促進し神経機能に障害を引き起こすことを発見しました。また、5-アミノレブリン酸がFXTASモデルマウスにおける神経機能障害の改善に有効であることを確認しました。難治性疾患であるFXTASの治療に新たな光を投げかける画期的な成果といえます。5-アミノレブリン酸は安全性の高い既承認医薬品であり、難病である「ATR-X症候群」のモデルマウスおよび患者における認知機能の低下に有効であることが既に示されています。
近年、FXTASと同様の症状を呈するCGGリピートに由来する疾患(トリプレットリピート病)が多数報告されています。また、アルツハイマー病やパーキンソン病等の神経変性疾患に、プリオノイドが関与することが示唆されています。これらの疾患に対して、今後、本研究により解明された「グアニン四重鎖によるプリオノイド神経障害メカニズム(G4プリオノイド)」を基盤とした神経変性疾患に対する治療法の開発が期待されます。
用語解説
- ※1 脆弱X随伴振戦/失調症候群(FXTAS;Fragile X-associated tremor/ataxia syndrome)
- X染色体に存在するFMR1遺伝子のCGGリピート配列が、子に受け継がれる内に次第に延長し発症するトリプレットリピート病の一種。正常なリピート数は45回以下だが、55~200回のリピートがあるとFXTASを発症し、中年以降から認知機能障害、精神症状やパーキンソン病様の症状を示す。
- ※2 グアニン四重鎖構造(G4)
- DNA、RNAの高次構造の一種。グアニンに富む核酸配列で形成される。4つのグアニンが四量体を作った面(G-カルテット)が2面以上重なった構造体。その構造特性からG4とも呼ばれる。
- ※3 プリオノイドタンパク質
- 牛海綿状脳症(BSE)やクロイツフェルト・ヤコブ病など、脳に異常なタンパク質が沈着し、脳神経細胞の機能が障害される病気(プリオン病)の病原体であるプリオンと同様な性質を有しているタンパク質のこと。正常タンパク質が異常タンパク質に変換し、細胞間伝播により病変を拡大するという学説をプリオノイド仮説と呼ぶ。
- ※4 RAN(Repeat-Associated Non-AUG)翻訳
- 一般的にタンパク質への翻訳は開始コドン(AUG)から始まる。しかし、特定の塩基配列の非常に長い繰り返し配列(リピート配列)が存在すると開始コドンがなくても翻訳が生じる。リピート配列の異常伸張に起因する多くの神経・筋疾患では、変性過程にある細胞にリピート配列由来のポリペプチドが蓄積しており、発症に重要な役割を果たしていると考えられている。
- ※5 液-液相分離(LLPS;liquid-liquid phase separation)
- 2つの液体が混ざり合わずに互いに排除しあうことで2相に分離する現象。細胞内では核酸やタンパク質がLLPSを起こして周囲とは異なる液相ができ、膜のないオルガネラとして液滴を形成する。LLPSは、ヘテロクロマチン形成、不溶性封入体の形成、細胞極性化など多くの細胞内現象に関与する。
参考文献
参考文献1;Shioda et al., Nature Medicine 24, 802-813. (2018)
参考文献2;Wada et al., Congenital Anomalies 60, 147-148. (2020)
参考文献3;Berman et al., J Neurodev Disord. 6, 25. (2014) Review
論文情報
- 論文名
- CGG repeat RNA G-quadruplexes interact with FMRpolyG to cause neuronal dysfunction in fragile X-related tremor/ataxia syndrome.
- 著者
- Sefan Asamitsu#, Yasushi Yabuki#, Susumu Ikenoshita, Kosuke Kawakubo, Moe Kawasaki, Shingo Usuki, Yuji Nakayama, Kaori Adachi, Hiroyuki Kugoh, Kazuhiro Ishii, Tohru Matsuura, Eiji Nanba, Hiroshi Sugiyama, Kohji Fukunaga, Norifumi Shioda*. (# Co-first authors; * Corresponding author)
- 掲載誌
- Science Advances
- DOI
- 10.1126/sciadv.abd9440
- URL
- https://advances.sciencemag.org/content/7/3/eabd9440
お問い合わせ先
熊本大学発生医学研究所 発生制御部門
ゲノム神経学分野
担当:准教授・塩田倫史(シオダノリフミ)
電話:096-373-6633
E-mail:shioda“AT”kumamoto-u.ac.jp
AMED事業に関するお問い合わせ先
難治性疾患実用化研究事業
日本医療研究開発機構 疾患基礎研究事業部 疾患基礎研究課
E-mail:nambyo-r“AT”amed.go.jp
※E-mailは上記アドレス“AT”の部分を@に変えてください。
関連リンク
掲載日 令和3年1月14日
最終更新日 令和3年1月14日