


ウイルス性脳炎はオートファジーの抑制により発症―ウエストナイルウイルスの病態発症機構を発見―
プレスリリース
北海道大学
日本医療研究開発機構
ポイント
- ウエストナイルウイルスのウイルスタンパク質が、細胞内のタンパク質の凝集体の形成を誘導。
- ウイルスタンパク質は、オートファジーに関連する宿主のタンパク質の分解を促すことが判明。
- オートファジーの抑制に注目することで、ウイルス性脳炎の新たな治療法の開発の進展が期待。
概要
北海道大学大学院獣医学研究院の小林進太郎助教、好井健太朗准教授(当時)らの研究グループは、世界中で流行し、重篤なウイルス性脳炎を引き起こすウエストナイルウイルス*1が、感染した神経細胞を傷害するメカニズムを明らかにしました。
多くのウイルスは、細胞内のタンパク質などを分解・除去するオートファジー*2により、増殖が抑制されることが知られています。本研究によりウエストナイルウイルスはオートファジーを抑制し、その結果、ウイルスに感染した細胞にタンパク質の凝集体の形成を誘導して、細胞死及び脳炎形成を引き起こすことが明らかになりました。更にウイルスが感染した細胞にオートファジーを誘導することでタンパク質の凝集体が除去され、細胞死が抑えられることも明らかになりました。多くのウイルス性脳炎は病気の発生メカニズムが分かっておらず、特異的な治療法もありません。本研究結果は特異的な治療法が存在しない多くのウイルス性脳炎に対し、病気の発生メカニズムや治療法の開発につながる成果であると考えられます。
本研究成果は、日本時間2020年1月24日(金)午前4時(米国太平洋時間2020年1月23日(木)午前11時)公開のPLOS Pathogens誌にオンライン公開される予定です。
なお本研究は、科学研究費補助金、国立研究開発法人日本医療研究開発機構(AMED)新興・再興感染症に対する革新的医薬品等開発推進研究事業、武田科学振興財団、独立行政法人日本学術振興会とチェコ科学アカデミーとの二国間交流事業(共同研究)により行われました。
背景
ウエストナイル脳炎は、蚊が媒介するウエストナイルウイルス(West Nile virus: WNV)によって引き起こされる人獣共通感染症*3の一つです。北米やヨーロッパを中心に世界中で発生が認められており、日本でも海外からの帰国者の感染が報告されています。
WNVは蚊の吸血によりヒトへと感染すると、末梢組織で一時的に増殖します。その後一部の感染者において、WNVは脳に移行し、神経細胞に感染し、細胞死及び重篤な脳炎を引き起こします。しかし、これまでにどのようなメカニズムで細胞死及び脳炎が引き起こされるのか、ほとんど明らかではありませんでした。
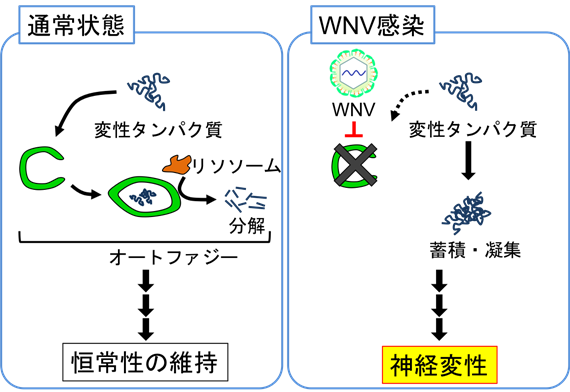
これまで小林助教らはWNVの病態モデルを構築し、WNVが感染している神経細胞に、アルツハイマー病などの神経変性疾患で蓄積が認められるタンパク質の凝集体が形成され、この形成が神経細胞死を誘導している可能性を示してきました(Neuropathology、 32:398-405)。正常な細胞ではこのようなタンパク質の凝集体は、オートファジーなどの細胞内タンパク質品質管理機構*4によって分解・除去されていることが知られています(参考図左)。そこで本研究では、オートファジーに着目して、WNVの感染によりタンパク質の凝集体の形成が起こるメカニズムを明らかにすることで、ウエストナイル脳炎の病態形成の機構について解明を目指しました。
研究手法
WNVのゲノムにコードされているウイルスタンパク質を神経系の培養細胞内に発現させることにより、タンパク質の凝集体の形成を誘導するウイルスタンパク質を特定しました。続いて、リバースジェネティクス法*5を用いて、タンパク質の凝集体を形成できないWNVを作製し、培養細胞やマウスモデルを用いて、WNVの感染がオートファジーに与える影響や病態形成への影響を解析しました。
研究成果
WNVのカプシドタンパク質*6の発現により、細胞内にタンパク質の凝集体が形成されることが明らかになりました。続いて、WNV感染細胞ではオートファジーが抑制されており、薬剤によるオートファジーの誘導によりタンパク質の凝集体が除去され、細胞死が抑えられることが明らかになりました。またWNV感染細胞では、オートファジーの誘導因子であるAMP-activated protein kinase(AMPK)*7の分解が亢進(こうしん)されており、カプシドタンパク質がAMPKと結合することも明らかになりました。タンパク質の凝集体の形成に重要なアミノ酸に変異を導入したWNVは、オートファジーの抑制やAMPKの分解を起こすことができず、マウスモデルにおける神経細胞の傷害や脳炎の発症が抑制されることが明らかになりました。以上の結果より、WNVはカプシドタンパク質によりオートファジーを抑制し、このことによるタンパク質の凝集体の蓄積が、中枢神経症状の発症に関与することが示されました(参考図右)。
- WNVの感染で起こるタンパク質の凝集体の形成には、WNVのカプシドタンパク質の特定のアミノ酸が重要であることが同定されました。
- カプシドタンパク質はオートファジーの誘導因子であるAMPKの分解を亢進(こうしん)することで、オートファジーを抑制することが明らかになりました。
- WNVの感染で起こるオートファジーの抑制やAMPKの分解は、脳炎の発症に関連することが明らかになりました。
今後への期待
オートファジーの異常はアルツハイマー病などの神経変性疾患など、さまざまな疾患の発症に関与することが明らかになってきています。今後、WNVの感染で起こるオートファジーの抑制機構が完全に明らかになることにより、ウイルス性疾患だけでなく、オートファジーの異常が関与するさまざまな疾患の病態の解明及び治療法の開発の契機になることが期待されます。
研究費・研究支援
本研究は、科学研究費補助金、国立研究開発法人日本医療研究開発機構(AMED)の新興・再興感染症に対する革新的医薬品等開発推進研究事業、武田科学振興財団、独立行政法人日本学術振興会とチェコ科学アカデミーとの二国間交流事業(共同研究)により行われました。
論文情報
- 論文名
- West Nile virus capsid protein inhibits autophagy by AMP-activated protein kinase degradation in neurological development(ウエストナイルウイルスはカプシドタンパク質によりAMP-activated protein kinaseを分解することでオートファジーを抑制し、神経病態を形成する)
- 著者名
- 小林 進太郎1、好井 健太朗1(当時)、3、Wallaya Phongphaew2、武藤 芽未1、 平野 港1、 大場 靖子2、 澤 洋文2、 苅和 宏明1(1北海道大学大学院獣医学研究院公衆衛生学教室、2北海道大学人獣共通感染症リサーチセンター分子病態診断部門、3長崎大学感染症共同研究拠点)
- 雑誌名
- PLOS Pathogens(微生物学の専門誌)
- DOI
- 10.1371/journal.ppat.1008238
- URL
- https://journals.plos.org/plospathogens/article?id=10.1371/journal.ppat.1008238
参考図
用語解説
- *1 ウエストナイルウイルス
- 蚊を介してヒトやウマなどの動物に感染し、発熱や脳炎を引き起こすウイルス。
- *2 オートファジー
- 細胞内のタンパク質を分解するための仕組みの一つであり、飢餓やウイルス感染などにより誘導される。
- *3 人獣共通感染症
- 同じ病原体により、ヒトとヒト以外の脊椎動物の双方が病気を発症する感染症のこと。
- *4 細胞内タンパク質品質管理機構
- 細胞内で合成に失敗したタンパク質を検出し、正しく機能するように調整したり、分解・除去したりする機構のこと。分解機構はオートファジーとプロテアソーム系に大きく分けることができる。
- *5 リバースジェネティクス法
- ウイルス学の分野において人工的に作製した遺伝子配列から、ウイルスを再産生する手法のこと。特定の遺伝子配列を変異させ、病原性を弱めたりすることができる。
- *6 カプシドタンパク質
- WNVのウイルス粒子を形成するタンパク質であり、細胞死を誘導することが報告されている。
- *7 AMPK
- 生体内のエネルギーセンサーであり、エネルギーが不足すると活性化され、オートファジーや脂質代謝を誘導する。
お問い合わせ先
研究内容に関すること
北海道大学大学院獣医学研究院
助教 小林 進太郎(こばやし しんたろう)
TEL:011-706-5213 FAX:011-706-5213
メール:shin-kobayashi"AT"vetmed.hokudai.ac.jp
北海道大学大学院獣医学研究科 環境獣医科学講座 公衆衛生学教室ウェブサイト
北海道大学大学院獣医学研究院
准教授 好井 健太朗(よしい けんたろう)
(現所属:長崎大学感染症共同研究拠点)
TEL:011-706-5212 FAX:011-706-5213
メール:kyoshii”AT”vetmed.hokudai.ac.jp
AMEDに関すること
国立研究開発法人日本医療研究開発機構(AMED)
戦略推進部 感染症研究課
TEL:03-6870-2225
メール:shinkou-saikou”AT”amed.go.jp
※メールは上記アドレス“AT”の部分を@に変えてください。
掲載日 令和2年1月24日
最終更新日 令和2年1月24日