


ALK融合遺伝子陽性肺がんに対する薬剤耐性克服薬の発見―第3世代ALK阻害薬耐性の克服を目指す―
プレスリリース
がん研究会
日本医療研究開発機構
ポイント
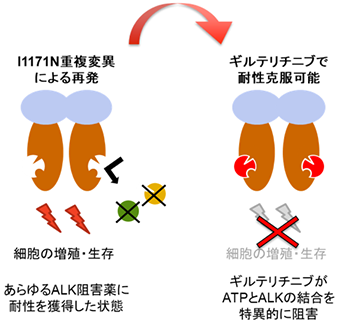
- ALK陽性肺がんにおいて、あらゆる既承認ALK阻害薬に耐性を示すALK-I1171N+F1174I及びI1171N+L1198H重複変異体は、急性骨髄性白血病の既承認薬であるギルテリチニブに感受性を示すことを発見しました。
- ギルテリチニブはALKを直接阻害し、様々な既存のALK阻害薬耐性変異体にも高い阻害活性を有することを発見しました。
- 最もALK阻害活性が高いと考えられているALK阻害薬ロルラチニブにも耐性は生じ、特にALK-L1198F変異の追加が高度耐性を誘導します。一方で、L1198F変異はギルテリチニブの感受性が亢進する変異でした。この理由についてスーパーコンピュータを用いたタンパク質構造シミュレーションにより、結合親和性の観点から明らかにすることができました。
- ROS1及びNTRK1融合遺伝子陽性の固形がんに対しても、ギルテリチニブは高い抗腫瘍効果を示しました。
- AXLを介したALK阻害薬耐性化機構にもギルテリチニブは有効であることを発見しました。
ALK陽性肺がんには5種類の承認された治療薬があり、1次治療に使用したアレクチニブなどのALK阻害薬に肺がんが耐性を示した場合には2次治療薬としてロルラチニブなどの他のALK阻害薬が使われます。このように耐性になったら次の異なるALK阻害薬を使用することで比較的長い期間の奏効が得られていますが、それでも複数の変異がALKに蓄積することで、あらゆるALK阻害薬に耐性が生じてしまいます。本研究において、これらの治療薬耐性を示すALK陽性肺がんに対し、急性骨髄性白血病治療薬のギルテリチニブが高い治療効果を示すことを明らかにしました。今後は臨床試験により更なる有効性を評価することで、新たな治療戦略となり得るか検証することが期待されます。
概要
我が国のがんによる死因の第1位は肺がんであり、その約85%が非小細胞肺がんに分類されます。この非小細胞肺がんの3~5%程度の患者さんでは、ALK融合遺伝子(注1)ががんの原因遺伝子として見つかります。ALK融合遺伝子を持つ肺がん(ALK陽性肺がん)に対しては、ALKに特異的な分子標的薬(ALKチロシンキナーゼ(注2)阻害薬[ALK阻害薬、注3])が有効です。現在我が国では、5つのALK阻害薬が承認されており、いずれも臨床試験で高い治療効果が示されてきました。しかし治療開始から数年以内に、薬剤耐性を獲得したがん細胞が出現し、がんが再発することが問題となっています。現在、実臨床では第2世代ALK阻害薬として知られるアレクチニブ(アレセンサ®)が第1選択薬として使用されることが多いですが、アレクチニブ治療後に現れる耐性メカニズムの約半分は、ALKキナーゼ部位における遺伝子変異が原因であることが報告されています。その耐性変異としては、G1202R(ALKの1202番目のアミノ酸であるグリシン(G)がアルギニン(R)に変異)変異やI1171N変異が高頻度で現れることが報告されています。このような耐性変異を有するがんにも第3世代ALK阻害薬のロルラチニブ(ローブレナ®)は有効であることが示されています。しかし、ロルラチニブに対してさえ、遺伝子変異がALKキナーゼに2つ以上入る重複変異により耐性が生じてしまうことが明らかとなっています。重複変異型ALKのいくつかはあらゆる既存のALK阻害薬に耐性を獲得することが判明しており、新たな治療戦略の開発が強く望まれていました(図1)。
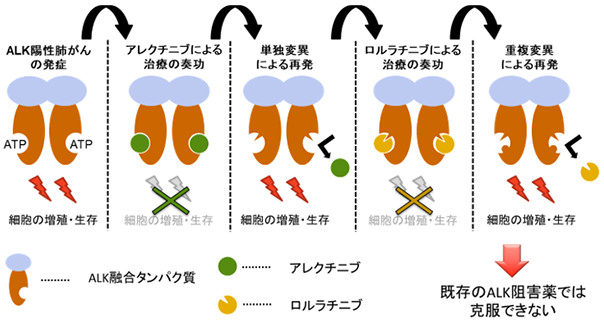
がん研究会の片山量平(がん研究会がん化学療法センター基礎研究部部長)、水田隼斗(慶應義塾大学大学院理工学研究科大学院博士課程)、岡田康太郎(東京大学大学院新領域創成科学研究科大学院博士課程)らの研究グループは、あらゆるALK阻害薬に耐性を示すI1171N+F1174I及びI1171N+L1198H重複変異体が、急性骨髄性白血病に対する既承認薬のギルテリチニブ(ゾスパタ®)に高い感受性を示すことを発見しました(図2)。
ALKの様に低頻度ながら肺がんにおいて融合遺伝子形成により強力ながん遺伝子として働いているROS1やNTRK1は、その活性中心であるチロシンキナーゼの構造がALKと非常に相同性が高いことが知られています。このROS1やNTRK1融合遺伝子陽性がんに対しても、ギルテリチニブは高い抗腫瘍効果を示すことを明らかにしました。
本研究の成果は、Nature Publishing Groupのオープンアクセス誌Nature Communicationsに、2021年2月24日午後7時(日本時間)に公開されます。
論文名、著者およびその所属
- 論文名
- Gilteritinib overcomes lorlatinib resistance in ALK-rearranged cancer
- ジャーナル名
- Nature Communications(Nature Publishing Groupのオープンアクセス誌)
(※2021年2月24日午後7時にオンラインに掲載されます。) - 著者
- Hayato Mizuta1,2†, Koutaroh Okada1,3†, Mitsugu Araki4, Jun Adachi5, Ai Takemoto1, Justyna Kutkowska1, Kohei Maruyama1,3, Noriko Yanagitani6, Tomoko Oh-hara1, Kana Watanabe7, Keiichi Tamai8, Luc Friboulet9, Kazuhiro Katayama10, Biao Ma11, Yoko Sasakura11, Yukari Sagae4, Mutsuko Kukimoto-Niino12, Mikako Shirouzu12, Satoshi Takagi1, Siro Simizu2, Makoto Nishio6, Yasushi Okuno4, Naoya Fujita1, Ryohei Katayama1,3*
(†筆頭著者、*責任著者) - 著者の所属機関
-
- 公益財団法人がん研究会 がん化学療法センター 基礎研究部
- 慶應義塾大学 理工学部 応用化学科
- 東京大学大学院 新領域創成科学研究科 メディカル情報生命専攻
- 京都大学大学院 医学研究科
- 国立研究開発法人医薬基盤・健康・栄養研究所
- 公益財団法人がん研究会 がん研有明病院 呼吸器内科
- 宮城県立がんセンター 呼吸器内科
- 宮城県立がんセンター 研究所 がん幹細胞研究部
- INSERM U981, Gustave Roussy Cancer Campus, Université Paris Saclay
- 日本大学 薬学部 薬学科
- 公益財団法人神戸医療産業都市推進機構 クラスター推進センター
- 理化学研究所 生命機能科学研究センター
研究の詳細
背景と経緯
我が国において肺がんは死亡者数1位のがん腫であり、年間約7万人以上の方が肺がんにより死亡しています。肺がんはさらに、非小細胞肺がんと小細胞肺がんに分類されますが、約85%を非小細胞肺がんが占めています。非小細胞肺がんの原因遺伝子としてはEGFRやKRAS遺伝子の異常が比較的高頻度に認められますが、3~5%の患者さんではALK融合遺伝子が強力ながん遺伝子として見つかります。受容体型チロシンキナーゼであるALKは、正常組織ではリガンドであるALKALが結合することで活性化し、細胞の増殖・生存シグナルは厳密に制御されています。しかし、EML4-ALKに代表されるALK融合遺伝子では、リガンド非依存的に恒常的なALKチロシンキナーゼの活性化が誘導され、増殖シグナルを異常に亢進させがん化へとつながります。ALKの異常な活性化がALK陽性肺がんの増殖を促進している本態であるため、ALK陽性肺がんには、分子標的薬であるALK阻害薬が有効です。現在までに、第1世代のクリゾチニブ、第2世代のアレクチニブ、セリチニブ、そして第3世代のロルラチニブが承認され、2021年1月にはブリグチニブが5剤目として、承認されました。第3相臨床試験の結果に基づき、第1選択薬としてはアレクチニブが広く使用されており、高い治療効果をもたらしています。しかし、薬剤耐性細胞の出現は避けられず、臨床上大きな問題となっています。アレクチニブ耐性化機構は主にALKの薬剤結合部位近傍における遺伝子変異によるアミノ酸置換の場合と、細胞内の他のキナーゼが活性化する場合の2つに分けられますが、前者の場合、ロルラチニブによる逐次治療が有効であることが示されています。しかしながら、同一ALK遺伝子内に2つ以上の変異が生じることで、ロルラチニブに耐性を示す重複変異の出現が実際の患者検体から発見され報告されており、今後より一層増加してくる可能性があります。重複変異体の一部には既存の第1、2世代ALK阻害薬が再び有効になる場合もありますが、一方でG1202R+L1196M重複変異体(ALKの1202番目のグリシン(G)がアルギニン(R)に変異し、同一遺伝子内の1196番目のロイシン(L)がメチオニン(M)に変異したもの)や、I1171N+F1174I重複変異体(ALKの1171番目のイソロイシン(I)がアスパラギン(N)に変異し、同一遺伝子内の1174番目のフェニルアラニン(F)がイソロイシン(I)に変異したもの)などはあらゆるALK阻害薬に耐性を示すため、新たな治療法の開発が強く望まれています。
がん研究会の片山量平(がん研究会がん化学療法センター基礎研究部部長)らを中心とする研究グループは、アレクチニブ-ロルラチニブ逐次治療後に出現する可能性の高い、あらゆるALK阻害薬に耐性を示す重複変異型ALKに対する新規克服薬の探索を目的として研究を行ってきました。
研究内容
IL-3依存的に増殖するマウス前駆Bリンパ球(Ba/F3細胞)にあらゆるALK阻害薬に耐性を示すI1171N+F1174I及びI1171N+L1198H重複変異型EML4-ALKをそれぞれ遺伝子導入し、変異型EML4-ALKに依存して増殖する細胞株を樹立しました。この細胞を用いて、既承認薬や臨床試験薬を中心に構成された阻害剤ライブラリーによるスクリーニングを行った結果、急性骨髄性白血病の既承認薬であるギルテリチニブが高い細胞増殖抑制効果を示しました(図3)。またALK陽性細胞株に対するギルテリチニブ処理時のリン酸化プロテオーム解析や、ALKの精製キナーゼを用いたin vitro kinase assayの結果、ギルテリチニブは確かにALKを直接阻害することが明らかになりました。
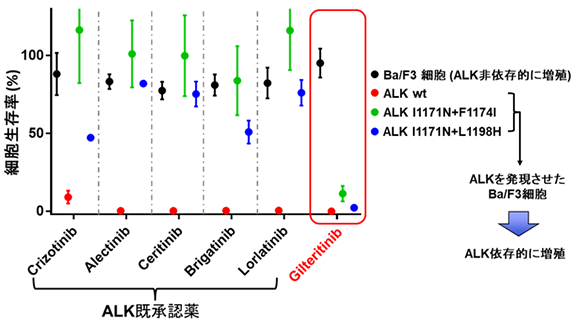
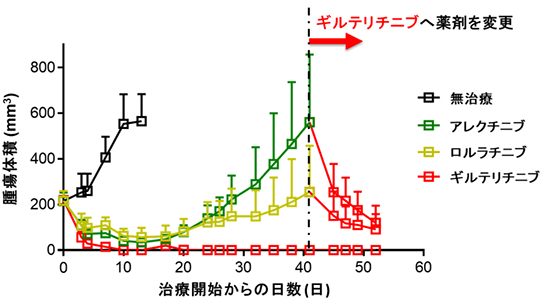
ALK阻害薬耐性変異体に対するギルテリチニブの阻害効果を検証するために、これまでに臨床及び前臨床で報告された耐性変異型EML4-ALKを導入したBa/F3細胞を用いて、細胞増殖試験、ウエスタンブロッティングを行った結果、G1202R、D1203N変異を除く単独変異と、I1171N/S重複変異型ALKが、ギルテリチニブに高い感受性を示すことを発見しました。そこでALK陽性患者由来細胞にALK-I1171N+F1174I変異体を導入した細胞株を樹立し、動物実験を行ったところ、既承認薬であるアレクチニブやロルラチニブは投与開始から20日ほどで腫瘍の再増大が観察された一方で、ギルテリチニブは50日以上腫瘍縮小効果が継続しました。また、アレクチニブやロルラチニブによる治療後に再増大してきた腫瘍も、ギルテリチニブへと変更することで、急速に腫瘍縮小する様子が観察されました(図4)。
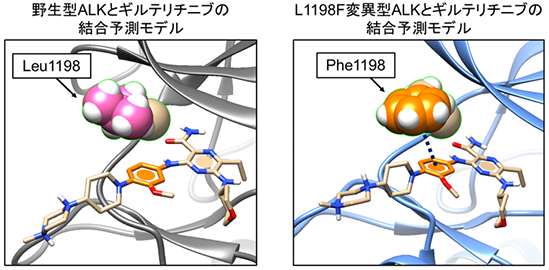
ギルテリチニブがどのようにALKに結合しているのかを詳細に解析するために、京都大学の奥野恭史(京都大学大学院医学研究科教授)、荒木望嗣(京都大学大学院医学研究科准教授)らの研究グループとの共同研究により、スーパーコンピュータを用いたタンパク質構造シミュレーションを行い、ギルテリチニブとの結合に重要なALKのアミノ酸残基・原子の推定に成功しました。細胞株を用いた実験から、ロルラチニブ耐性を誘導するL1198F変異は、野生型ALKよりもギルテリチニブに高い感受性を示すことを見出していましたが、コンピュータシミュレーションによる結合予測を行ったところ、1198番目のロイシンがフェニルアラニンに変異することで、側鎖のフェニル基がギルテリチニブとπ電子間相互作用することにより結合が安定化し、薬剤感受性を高めることが示唆されました(図5)。
また、ALK遺伝子変異を伴わない耐性化機構に対するギルテリチニブの有効性を検証するために、EGFR活性化によりALK阻害薬に耐性化した患者由来細胞や、活性化型KRAS(KRAS G12C変異)を遺伝子導入したALK陽性肺がん細胞を用いた実験を行いました。その結果、ギルテリチニブ単剤では耐性を克服できなかったものの、それぞれEGFR阻害薬やKRAS G12C阻害薬との併用療法により、耐性克服が可能になりました。一方で、肺がんを含む様々ながん腫において、治療残存腫瘍細胞(注4)の出現に寄与することが示唆されているAXL(注5)の活性化も、耐性化機構の1つとして挙げられます。実際に細胞レベルでは、ALK阻害薬に耐性を示すALK陽性肺がん細胞では、AXLの発現増加が報告されています。ギルテリチニブはAXLをも阻害することが知られていたため、AXLを遺伝子導入したALK陽性肺がん細胞株を樹立し、動物実験によりアレクチニブとギルテリチニブの抗腫瘍効果を比較しました。その結果、アレクチニブはAXLを発現した腫瘍の再増大を抑制できませんでしたが、ギルテリチニブでは同投与期間において腫瘍の再増大は観察されず、アレクチニブからギルテリチニブにスイッチすることで再び腫瘍増殖抑制が認められました。
ALKと比較的高い相同性を有する受容体型チロシンキナーゼROS1、NTRK1もALKと同様に融合遺伝子を形成し、肺がんなどの原因遺伝子になることが報告されており、それぞれ非小細胞肺がんの1%、0.1%を占めるといわれています。そこでROS1、NTRK1融合遺伝子を有する肺がん、大腸がんに対するギルテリチニブの抗腫瘍効果を動物実験で検証したところ、薬剤非投与群と比較して有意な腫瘍縮小効果が明らかになりました。
本研究により新規ALK薬剤耐性克服薬としてギルテリチニブの可能性が示されました。ギルテリチニブは急性骨髄性白血病に対して承認されている剤ますが、肺がん患者へ投与した際の安全性やALK陽性肺がんへの有効性を明らかにするためには、臨床試験による検討が必須です。
本研究への支援
本研究は、下記機関より資金的支援等を受けて実施されました。
- 国立研究開発法人日本医療研究開発機構(AMED) 次世代がん医療創生研究事業(P-CREATE)
「異分野先端技術融合による薬剤抵抗性を標的とした革新的複合治療戦略の開発」 - 独立行政法人日本学術振興会 科学研究費補助金
- 日本財団
- 公益財団法人 上原記念生命科学財団
- 文部科学省 ポスト「京」で重点的に取り組むべき社会的・科学的課題に関するアプリケーション開発・研究開発 重点課題1(課題責任者 奥野恭史 教授)
- 本論文の一部は、理化学研究所のスーパーコンピュータ「京」を利用して得られたものです。(課題番号:p160213、hp150272、hp170275、hp180186、ra000018)
用語解説
- (注1)ALK融合遺伝子
- 通常、ALK遺伝子をもとに作られるALKタンパク質は細胞膜上に発現するチロシンキナーゼ受容体であり、胚発生時の神経システムの発達に関わっています。増殖因子(ALK-AL)が結合することで、細胞増殖シグナルを活性化し、細胞の生存・増殖を促します。しかしながら、染色体逆位や転座により、ALK遺伝子がEML4遺伝子に代表されるような多量体化能を有する他の遺伝子と融合遺伝子を形成する場合があります。このALK融合遺伝子から作られるALK融合タンパク質は、増殖因子の有無にかかわらず恒常的にALKからの増殖シグナルを異常に活性化するため、がん化を誘導します。
- (注2)チロシンキナーゼ
- キナーゼとは基質をリン酸化する酵素の総称であり、そのうちチロシンキナーゼは基質タンパク質のチロシン残基をリン酸化する酵素のことです。一般にその活性化は私たちの細胞の増殖を正に誘導します。
- (注3)ALK阻害薬
- 現在、我が国においては、クリゾチニブ、アレクチニブ、セリチニブ、ブリグチニブが1次治療やそれ以降の治療において使用することができ、ロルラチニブはこれらの阻害薬耐性後の治療薬として使用されています。基質をリン酸化する際に必須となるATPとALKとの結合を阻害することでALKの活性化を抑制しています。
- (注4)治療残存腫瘍細胞
- がん細胞を標的とした薬剤を投与することで大部分のがん細胞は死滅し、腫瘍の退縮が確認されます。しかしCT検査では捉えきれない様なごくわずかながん細胞は、死滅することなく生体内で生存し続けます。一方で薬剤耐性細胞とは異なり、薬剤存在下で急激に増殖することもないと考えられています。長期間、薬剤存在下で生存する過程において、投与されている薬剤の標的とは異なるキナーゼが活性化したり、耐性変異を獲得したりすることで、薬剤耐性細胞へと進化し、がん再発へとつながると考えられており、いわば薬剤耐性細胞の芽となるような細胞です。
- (注5)AXL
- AXLはALKと同じく受容体型チロシンキナーゼの1つで、慢性骨髄性白血病患者より初めて単離、精製されました。通常リガンドであるGAS6の結合に依存して活性化し、細胞の増殖・生存に関わります。一方で一部のがん細胞ではAXLの異常な発現上昇に伴い、リガンド非依存的に異常な増殖・生存シグナルの活性化がみられます。さらに近年、AXLの活性化が、細胞を上皮系から間葉系へと形質を変化させたり(様々な細胞内因子の発現量を変化させ、上皮様(敷石状)の形態をした細胞が細長い見た目(間葉系)の細胞となる現象でEMT(上皮間葉転換)と呼ばれます)、AXLとAXL以外のキナーゼが結合して、増殖・生存シグナルを活性化させたりすることで、薬剤存在下でのがん細胞の生存を助けることが示唆されています。
お問い合わせ先
本研究に関すること
公益財団法人がん研究会 がん化学療法センター 基礎研究部
片山量平
(〒135‐8550 東京都江東区有明3-8-31)
TEL:03-3520-0111
取材等に関すること
公益財団法人がん研究会 広報部
TEL:03-3570-0775
がん対策全般についてのお問い合わせ
厚生労働省健康局 がん・疾病対策課
(〒100-8916 東京都千代田区霞が関1-2-2)
TEL:03-5253-1111(内線4605)
次世代がん医療創生研究事業に関するお問い合わせ
国立研究開発法人日本医療研究開発機構(AMED)
創薬事業部 医薬品研究開発課
(〒100-0004 東京都千代田区大手町一丁目7番1号)
TEL:03-6870-2311
E-mail:cancer“AT”amed.go.jp
※E-mailは上記アドレス“AT”の部分を@に変えてください。
関連リンク
掲載日 令和3年2月25日
最終更新日 令和3年2月25日