


多発性硬化症治療薬が作用する受容体の構造基盤を解明―副作用の少ない安全性の高い薬剤の開発に貢献―
プレスリリース
東北大学大学院薬学研究科
日本医療研究開発機構
発表のポイント
- 多発性硬化症(注1)治療薬と結合した生理活性脂質スフィンゴシン1-リン酸(S1P、注2)受容体の立体構造を解明しました。
- 多発性硬化症治療薬結合時に特徴的なS1P受容体の内部の構造変化を見出し、S1P受容体を分解に導く構造基盤を明らかにしました。
- 本研究は、受容体の機能(シグナル伝達や分解)を選択的に誘導する薬剤、すなわち副作用の少ない安全性の高い創薬開発に貢献する研究成果です。
概要
自己免疫性疾患の一種である多発性硬化症の患者では、自己反応性の異常Tリンパ球(注3)がS1Pに応答して体内を循環し、脳に到達して過剰な自己免疫反応を引き起こします。多発性硬化症治療薬であるフィンゴリモド(商品名イムセラ)やシポニモド(商品名メーゼント)は、自己反応性Tリンパ球の表面上に存在するS1P受容体(S1PR1)に作用します。治療薬が結合したS1PR1は、S1Pが結合した場合と異なり、細胞内への持続的な取り込みとそれに引き続く分解を受けます。従って、多発性硬化症治療薬を服用した患者の自己反応性Tリンパ球はS1Pに応答できず、脳への移行が阻害されて炎症が抑制されることが知られています(図1)。一方これら治療薬がS1PR1に対してどのような構造変化を誘導することで、分解経路に至るのかその機構は明らかになっていませんでした。
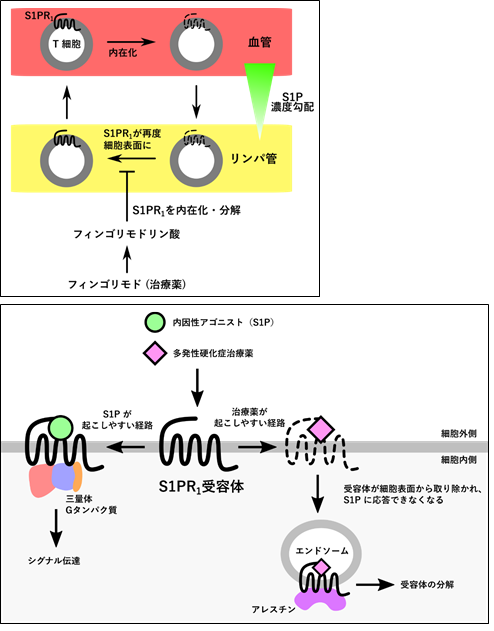
Tリンパ球は血中とリンパ中を循環しており、リンパ管から血中への移行する際にS1Pの濃度勾配をTリンパ球の細胞表面に存在するS1PR1が感知する。フィンゴリモドは体内で代謝活性体のリン酸化型となると、Tリンパ球のS1PR1に結合してこれを分解に導く。多発性硬化症患者においては、自己反応性の異常Tリンパ球がリンパ中から血中へ移行できず、脳における自己炎症が抑制される。
(下)多発性硬化症治療薬のアレスチン(注4)バイアス効果
S1PR1は三量体Gタンパク質(注5)を介したシグナル伝達(細胞遊走など)とアレスチンを介した内在化の応答を引き起こす。フィンゴリモドを始めとする多発性硬化症治療薬はS1Pと比べてアレスチン経路を誘導しやすいことから、アレスチンバイアスリガンドとして知られる。
東北大学大学院薬学研究科の井上飛鳥准教授らの研究グループは、中国ハルビン工業大学のYuanzheng He教授らのグループとの共同研究により、3種類の化合物(S1P、フィンゴリモドの代謝活性体、シポニモド)がそれぞれ結合したS1PR1の立体構造を解明し、それに基づいた分子動力学シミュレーション(注6)と機能解析を行うことで治療薬結合型受容体の作動基盤を解明しました。今回の研究成果は、S1PR1を標的にする薬効に優れた多発性硬化症治療薬の開発に貢献するとともに、S1P受容体以外のGタンパク質共役受容体(GPCR、注7)においても副作用を低減させた薬剤の開発に役立つことが期待されます。
本研究の成果は、日本時間2021年12月23日に科学雑誌Nature Chemical Biology誌の電子版に掲載されました。
詳細な説明
研究の背景
スフィンゴ脂質の一種であるS1Pは、前駆体であるスフィンゴシンが細胞内でスフィンゴシンキナーゼによってリン酸化されることで生合成されます。S1Pは輸送体によって細胞外に放出されると、近傍の細胞の表面に存在するGPCRの一種であるS1P受容体に結合・活性化することで、下流シグナル因子である三量体Gタンパク質のシグナル伝達を誘導します。S1P受容体は5種類(S1PR1-S1PR5)存在しますが、なかでもS1PR1は免疫に関わるTリンパ球に高発現していることが知られています。Tリンパ球は血管とリンパ管を循環して体内の異物を監視しており、リンパ節から血中への移行の際には、S1Pの濃度勾配によるTリンパ球の遊走が必須であることが知られています。血中の高濃度のS1Pに晒されるとTリンパ球のS1PR1は細胞内へと内在化し、S1P濃度の低いリンパ管に移行してS1PR1が再び細胞表面に出てくるまでS1Pに不応答となります(図1上)。免疫抑制剤として開発された薬剤であるフィンゴリモドは、体内に取り込まれるとS1Pと同様にスフィンゴシンキナーゼによって代謝活性体であるリン酸化体に変換されます。このフィンゴリモドリン酸はS1Pと同様にS1PR1に結合し(結合分子を一般にリガンドと呼びます)、三量体Gタンパク質のシグナル伝達を誘導します。一方で、フィンゴリモドリン酸はS1PR1をS1Pよりも強く内在化させ、最終的に細胞内で分解させることによって、Tリンパ球の体内循環を阻害して免疫を抑制すると考えられています(図1上)。S1PR1を含むGPCRはアレスチンと呼ばれるシグナル調節タンパク質と結合することで内在化されることが知られています。フィンゴリモドリン酸が結合したS1PR1は三量体Gタンパク質よりもアレスチンを好むことから、アレスチンバイアスリガンド(アレスチンに「偏る」結合因子の意)と呼ばれています。また、2020年に本邦で多発性硬化症の治療薬として認可されたシポニモドもアレスチンバイアスリガンドであることが知られています(図1下)。一方で、これらの治療薬がなぜバイアスを引き起こすのかは不明なままであり、GPCR一般に対するバイアスの詳細な機序も解明されていませんでした。
研究の内容
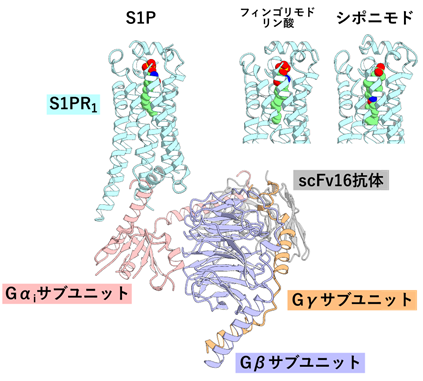
今回、東北大学大学院薬学研究科の生田達也助手・井上飛鳥准教授らの研究グループは、中国ハルビン工業大学のYuanzheng He教授らのグループとの共同研究により、S1Pやフィンゴリモドリン酸、シポニモドが結合したS1PR1および三量体Gタンパク質で構成されるシグナル伝達複合体の立体構造を、クライオ電子顕微鏡を用いて決定しました(図2)。そして、これらの構造に基づいたS1PR1の1アミノ酸変異体の機能解析により、リガンドの認識機構を明らかにしました(図3)。また、これらの構造に基づく分子動力学シミュレーションによって、アレスチンバイアスリガンドに特有のS1PR1の内部の構造変化を見出しました。さらに、特定された構造変化に関わるアミノ酸残基を置換したS1PR1変異体を作製し、三量体Gタンパク質の活性化とS1PR1とアレスチン複合体の内在化を測定することで、見出された構造変化がバイアスに関わることを実証しました(図4)。
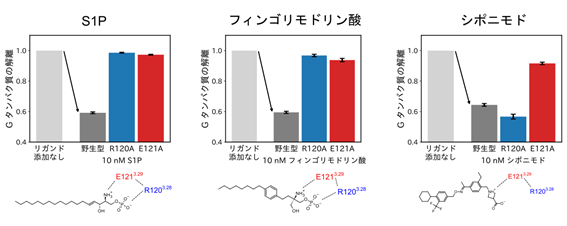
S1PR1に対するリガンド結合を三量体Gタンパク質の活性化(乖離)応答として評価した。野生型S1PR1は3種類のリガンドのいずれに対しても応答し、三量体Gタンパク質の乖離を誘導した(矢印)。一方、R120A変異体(青)はシポニモドに応答したが、S1Pとフィンゴリモドリン酸に対する応答が消失した。E121A変異体(赤)は全てのリガンドに対して不応答になった。
(下)各リガンドの認識機構の模式図
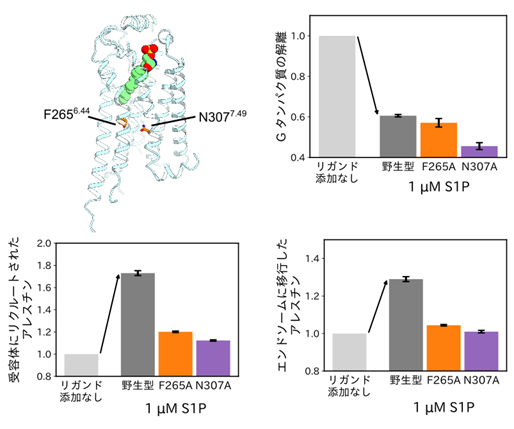
リガンド結合部位からやや離れた位置に存在するアミノ酸残基(F265とN307)がアレスチンバイアスリガンド結合時に相互作用することが、分子動力学シミュレーション計算から判明した。
(右上、下)アミノ酸残基をアラニンに置換したS1PR1変異体の各経路(図1参照)の測定結果。
F265AとN307Aの各変異体はGタンパク質に対する活性は野生型S1PR1と同等であるものの(右上)、アレスチンを結合する活性(左下)とエンドソーム(注8)に移行する活性(右下)は野生型S1PR1と比べて大幅に減弱した。
GPCRはリガンドが細胞外から結合するとその立体構造が変化し、細胞内にGα/Gβ/Gγの3つのサブユニットからなる三量体Gタンパク質を結合します。その後、三量体Gタンパク質は核酸交換反応を経て、GαサブユニットとGβγサブユニットに解離します。本研究グループは、三量体Gタンパク質の改変体を用いてこの解離を測定し、S1PR1のGタンパク質経路を測定しました。S1PR1とともに分割ルシフェラーゼ(注9)を融合したGαサブユニットとGγサブユニットを培養細胞に発現させると、ルシフェラーゼ由来の発光が検出できます。この状態で、S1PR1リガンドを添加してGタンパク質が活性化されると、発光の減弱を継時的に測定できます。この評価系を用いて、種々の変異を導入したS1PR1の三量体Gタンパク質シグナルを評価しました。リガンド結合ポケットのアミノ酸残基を変異させて(120番目のアルギニン(R120)と121番目のグルタミン酸(E121)をそれぞれアラニンに置換)、三量体Gタンパク質の活性化を測定したところ、S1Pとフィンゴリモドリン酸は類似の認識機構によってS1PR1に結合する一方、シポニモドは別のアミノ酸残基を介した認識機構によってS1PR1に結合することがわかりました(図3)。
これら3種のリガンドが結合したS1PR1のクライオ電顕構造は、全体として極めて似通った構造をとっておりバイアス性がどのように生じるか不明でした。そこで、分子動力学シミュレーションと呼ばれる計算機シミュレーションを行い、運動方程式に従った原子の経時変化を追跡しました。このシミュレーションを異なるリガンドが結合した状態のS1PR1に対して行い、それぞれの結果を解析・比較しました。その結果、アレスチンバイアスリガンドであるフィンゴリモドリン酸とシポニモドに共通し、S1Pとは異なるS1PR1の内部のアミノ酸残基同士の相互作用を見出しました。そこで、この残基を変異させたS1PR1がどのようなバイアスを示すか実験で確かめました(図4)。上述の三量体Gタンパク質経路に関する測定に加え、細胞膜やエンドソームにおけるアレスチンとS1PR1の複合体の局在をルシフェラーゼ断片の相互作用によって評価しました。その結果、着目した相互作用(265番目のフェニルアラニン(F265)と307番目のアスパラギン(N307)、図4)を低下させたアラニン変異体では、三量体Gタンパク質とアレスチンのバランスが変化することを確認でき、これらの相互作用がS1PR1機能のバイアス性の起因であることを明らかにしました。
今後の展望
本研究は、S1PR1に対する薬剤の結合様式を示し、今後のS1PR1に対する薬剤開発に役立つと期待されます。また、アレスチンバイアスリガンド特有の受容体内部の相互作用はGPCRによく保存された残基が関わるため、S1P受容体に限らず他の多くのGPCRに対しても適用可能であることが期待されます。
本研究は、日本学術振興会(JSPS)の科学研究費助成事業(21H04791、21H051130)や国立研究開発法人日本医療研究開発機構(AMED)の革新的先端研究開発支援事業ソロタイプ(PRIME、JP19gm5910013)、インキュベートタイプ(LEAP、JP20gm0010004)、創薬等ライフサイエンス研究支援基盤事業(BINDS、JP20am0101095)、国立研究開発法人科学技術振興機構(JST)のムーンショット型研究開発事業(JPMJMS2023)、第一三共生命科学研究振興財団、武田科学振興財団、小野医学研究財団、上原記念生命科学財団など多くの支援を受けて行われました。
用語説明
- (注1)多発性硬化症
- 免疫系によって自身の神経細胞を包むミエリンが破壊され炎症が起こる疾患であり、再発と寛解を繰り返すことで炎症部位が硬くなることに由来して名付けられた。特定疾患に指定されている神経難病。
- (注2)スフィンゴシン1-リン酸(S1P)
- 細胞の脂質二重層を形成するスフィンゴ脂質の代謝物であり、スフィンゴシンがリン酸化されてS1Pとなった後に細胞外へと放出され、細胞間のシグナル分子として働く。
- (注3)Tリンパ球
- リンパ節中に多い白血球の一種。Bリンパ球が抗体を産生するのに対し、Tリンパ球は標的を直接攻撃することができる。今回の研究対象であるS1PR1を高発現する。
- (注4)アレスチン
- Gタンパク質と競合してGPCRと相互作用するタンパク質であり、GPCRの内在化を促進するタンパク質の足場となる。
- (注5)三量体Gタンパク質
- Gα/Gβ/Gγの3つのサブユニットからなるタンパク質複合体であり、GPCRによってGTP結合型GαとGβγに解離する。解離後のサブユニットは下流のシグナル分子と相互作用することによって、さまざまな細胞内シグナル応答を引き起こす。
- (注6)分子動力学シミュレーション
- 計算機を用いたシミュレーション手法のひとつであり、原子同士に働く相互作用力に基づいて運動方程式を計算することで原子位置の変化を見ることができる。
- (注7)Gタンパク質共役(きょうやく)受容体(GPCR)
- 細胞表面に存在する受容体であり、細胞外のシグナル分子と結合し、細胞内の三量体Gタンパク質を介したシグナル伝達を介在する。7回細胞膜を貫通する特徴的な構造を有する。ヒトでは約300種類のGPCRが存在し、約3割の既存薬がいずれかのGPCRと結合して薬効を発揮することが知られる。
- (注8)エンドソーム
- 細胞膜の一部が凹んで形成される小胞の一種。エンドソームから細胞表面に戻る輸送経路とタンパク質分解に至る経路が分かれる。
- (注9)分割ルシフェラーゼ
- 生体発光反応を触媒する酵素(ルシフェラーゼ)を分割した断片であり、別々のタンパク質に融合することでタンパク質間の相互作用の測定に利用できる。本研究ではNanoLucと呼ばれる深海エビ由来のルシフェラーゼをもとに作られたSmBiT、LgBiTと呼ばれる2断片を用いた。
論文情報
- 雑誌名
- Nature Chemical Biology
- 論文タイトル
- Structural basis of sphingosine-1-phosphate receptor 1 activation and biased agonism
- 著者
- Zhenmei Xu†, Tatsuya Ikuta†, Kouki Kawakami, Ryoji Kise, Yu Qian, Ruixue Xia, Ming-Xia Sun, Anqi Zhang, Changyou Guo, Xue-Hui Cai, Zhiwei Huang, Asuka Inoue*, Yuanzheng He*(†:共同筆頭著者、*:共同責任著者)
- DOI番号
- 10.1038/s41589-021-00930-3
- アブストラクトURL
- http://dx.doi.org/10.1038/s41589-021-00930-3
お問い合わせ先
研究内容に関すること
東北大学大学院薬学研究科
准教授 井上飛鳥
Tel:022-795-6861
E-mail:iaska“AT”tohoku.ac.jp
AMED事業に関すること
日本医療研究開発機構(AMED)
シーズ開発・研究基盤事業部 革新的先端研究開発課
Tel:03-6870-2224
E-mail:kenkyuk-ask“AT”amed.go.jp
※E-mailは上記アドレス“AT”の部分を@に変えてください。
関連リンク
掲載日 令和3年12月24日
最終更新日 令和3年12月24日