


炎症細胞によるがん転移性ニッチ形成メカニズムを解明
プレスリリース
国立研究開発法人日本医療研究開発機構
発表者
発表のポイント
- Mint3が炎症性モノサイトの機能制御を介してがん転移性ニッチの形成を制御していることを明らかにした。
- 炎症細胞によるがん細胞の血管外遊走制御の新たなメカニズムが解明された。
- 炎症細胞のMint3を標的とすることでがんの転移を阻害する薬剤の開発が期待される。
発表概要
東京大学医科学研究所 癌・細胞増殖部門 人癌病因遺伝子分野の坂本毅治助教らの研究グループは、炎症細胞によるがん転移の促進に関わる重要な分子として、Mint3を発見しました。
局所で増殖するがんには外科手術や放射線治療などが有効ですが、がんが全身に転移すると有効な治療法があまりないため、がんの転移を抑える薬剤の開発が待望されています。がん細胞が遠隔臓器に転移するには、原発巣のがん細胞が血管内に入り、転移先臓器で血管外へ遊走する必要があります。がん細胞の血管外遊走にはがん細胞だけでなく炎症細胞などの助けが必要なことが分かっていましたが、その詳細なメカニズムは不明でした。本研究では、Mint3という分子が、炎症細胞の一種である炎症性モノサイト(注1)の機能を制御することで、がん細胞が転移先臓器で血管外遊走しやすくなる転移性ニッチ(注2)を形成していることを明らかにしました。本研究の成果によって、炎症細胞のMint3を標的としたがん転移阻害薬の開発が期待されます。
本研究成果は2017年5月15日(米国東部時間 午後3時)、米国科学雑誌「Proceedings of the National Academy of Sciences of the United States of America」のオンライン速報版で公開されます。
本研究成果は、日本医療研究開発機構(AMED)次世代がん研究シーズ戦略的育成プログラム(P-DIRECT)(平成23年度~平成27年度)、次世代がん医療創生研究事業(P-CREATE)(平成28年度以降)、文部科学省科学研究費補助金新学術領域研究などの一環として得られました。
発表内容
研究の背景・先行研究における問題点
研究内容
本研究では、Mint3という分子が、炎症細胞の一種である炎症性モノサイトの機能を制御することで、がん細胞が転移先臓器で血管外遊走しやすくなる転移性ニッチの形成を制御していることを明らかにしました。
まず、Mint3遺伝子を欠損したマウスを用いて、がん細胞をマウス尾静脈に移植し肺への転移を観察したところ、Mint3欠損マウスでは、野生型のマウスに比べ、移植したがん細胞の肺への転移が有意に抑制されていました(図1)。また、Mint3欠損マウスでは、がん細胞が血管内に留まったまま転移先臓器に移動できず、最終的に排除されることが明らかとなりました。つまり、Mint3はがん転移を促進することが分かりました。
さらなる解析により、Mint3によるがん転移の促進には炎症性モノサイトが重要な役割を果たしていることが分かりました。Mint3は低酸素誘導因子HIF-1と呼ばれる転写因子(注3)を活性化することが知られています。本研究では、Mint3が炎症性モノサイトのHIF-1を活性化することで、1. 炎症性モノサイトの転移臓器への集積、2. VEGF(血管内皮増殖因子、注4)発現の促進、の2種類のメカニズムを介して、転移性ニッチの構築を行うことを明らかにしました(図2)。
メカニズム1:炎症性モノサイトで、Mint3はHIF-1の標的遺伝子である解糖系(注5)関連遺伝子の発現を増加させます。解糖系が働くことで、炎症性モノサイトが遠く離れた臓器まで移動するのに必要なエネルギーが供給されます。Mint3欠損マウスでは、炎症性モノサイトの解糖系がうまく働かずエネルギー不足で転移先臓器に移動出来ず、その結果、転移先臓器に集まった炎症性モノサイトの数が減少していました。
メカニズム2:Mint3は炎症性モノサイトでHIF-1の標的遺伝子であるVEGFの発現も促進します。転移先臓器で、炎症性モノサイトからVEGFが産生されると、血管内皮細胞同士の結合が緩くなり(血管透過性の亢進)、さらに血管内皮細胞にE-selectinという接着分子(注6)の発現が誘導されます。がん細胞は血管内皮細胞のE-selectinを利用して血管内皮細胞に接着し、そして血管内皮細胞の隙間を通り抜けることにより、転移先臓器への血管外遊走を起こします(図3)。Mint3欠損マウスでは、炎症性モノサイトのVEGF産生が低下しているため、転移先臓器のE-selectinの発現誘導と血管透過性が低下していました。
以上、Mint3が炎症性モノサイトの転移先臓器での数(量)と機能(質)の両方を制御することでがん転移性ニッチを形成していることが本研究により明らかとなりました。
社会的意義
発表雑誌
論文タイトル:Control of metastatic niche formation by targeting APBA3/Mint3 in inflammatory monocytes
著者:Toshiro Hara, Hiroki J. Nakaoka, Tetsuro Hayashi, Kouhei Mimura, Daisuke Hoshino, Masahiro Inoue, Fumitaka Nagamura, Yoshinori Murakami, Motoharu Seiki, and Takeharu Sakamoto*
用語解説
- (注1)炎症性モノサイト:
- 血液中にいる炎症細胞の1種で、炎症の誘導や制御に関わる。
- (注2)転移性ニッチ:
- がんが転移しやすくなるような生体内の環境。
- (注3)転写因子:
- DNA上の特定の配列に結合することで標的遺伝子の発現量を変化させる分子。
- (注4)VEGF:
- 血管が新たに作られる際に分泌されるタンパク。血管内皮細胞に作用し、細胞増殖、血管透過性の亢進、接着分子の発現などを誘導する。
- (注5)解糖系:
- 細胞外から取り込んだグルコースを細胞質内の一連の酵素反応で代謝することでエネルギー(ATP)を産生する代謝経路。
- (注6)接着分子:
- 細胞表面にあるタンパクで、他の細胞の接着分子やコラーゲンなどの足場タンパクと結合することで、細胞が他の細胞や足場に接着できるようにする働きがある。E-selectinは接着分子の1種。
添付資料
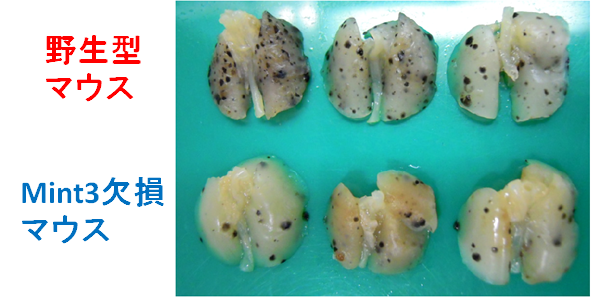
図1:マウスメラノーマB16F10細胞の肺への転移
Mint3欠損マウスでは野生型マウスに比べてB16F10細胞の肺の転移結節(黒色の塊)が減少している。
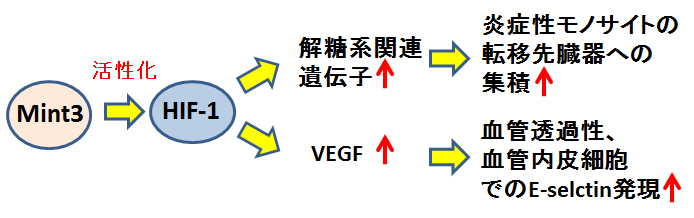
図2:Mint3の炎症性モノサイトでの役割
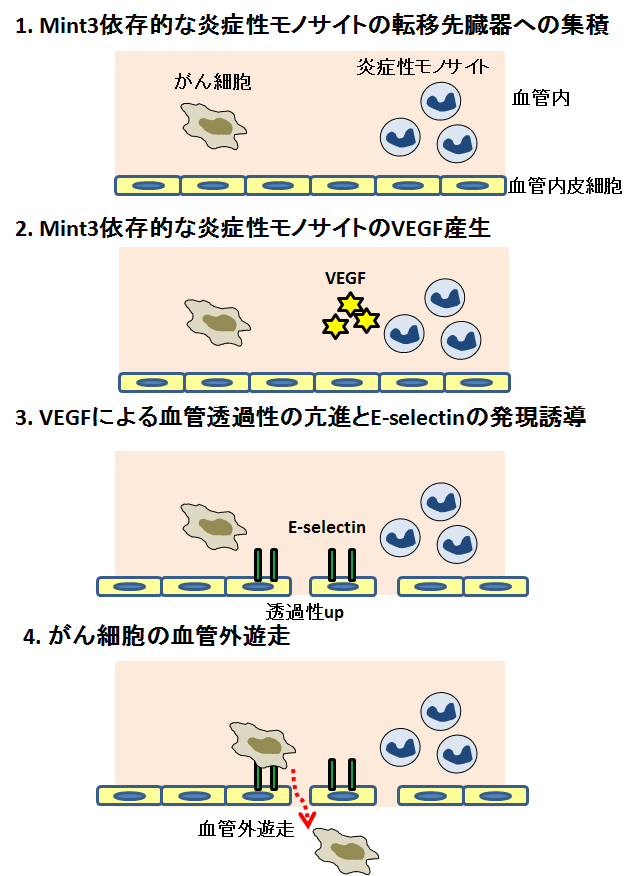
図3:Mint3による転移性ニッチの形成
お問い合わせ先
研究に関するお問い合わせ
東京大学医科学研究所 癌・細胞増殖部門 人癌病因遺伝子分野
助教 坂本 毅治(サカモト タケハル)
TEL:03-5449-5264
FAX:03-5449-5407
E-mail:t-saka“AT”ims.u-tokyo.ac.jp
報道に関するお問い合わせ
東京大学医科学研究所 国際学術連携室
TEL:03-6409-2027
E-mail:koho“AT”ims.u-tokyo.ac.jp
国立研究開発法人日本医療研究開発機構(AMED)
経営企画部 企画・広報グループ
Tel:03-6870-2245
AMED の事業に関するお問い合わせ
国立研究開発法人日本医療研究開発機構(AMED)
戦略推進部 がん研究課
次世代がん医療創生研究事業担当
〒100-0004 東京都千代田区大手町1-7-1
TEL:03-6870-2221
FAX:03-6870-2244
E-mail:cancer“AT”amed.go.jp
※Emailは上記アドレス“AT”の部分を@に変えてください。
掲載日 平成29年5月16日
最終更新日 平成29年5月16日