


脳腫瘍に対するウイルス療法の医師主導治験で高い治療効果を確認―日本初のがん治療ウイルス薬の製造販売承認申請へ―
プレスリリース
国立大学法人東京大学
国立研究開発法人日本医療研究開発機構
東京大学医科学研究所附属病院 脳腫瘍外科 藤堂具紀教授らの研究グループは、単純ヘルペスウイルス1型(口唇ヘルペスのウイルス)に人工的に3つのウイルス遺伝子を改変(三重変異)した第三世代のがん治療用ヘルペスウイルス G47Δ(ジーよんじゅうななデルタ)を用いた、膠芽腫(こうがしゅ、悪性脳腫瘍の一種)の患者を対象にした医師主導治験において、中間解析の結果、G47Δの高い治療効果を確認しました。本治験は、治療効果の検討を目的とした第Ⅱ相臨床試験(注1)で、再発もしくは残存した膠芽腫病変に対し、最大6回の腫瘍内投与を行ったところ、治療開始から1年経過した患者13名において、主要評価項目である一年生存割合(治療開始後1年間生存した患者の割合)が92.3%であり、他の複数の臨床試験結果から算出された標準治療の一年生存割合(15%)と比較して高い有効性を示しました。一方、G47Δ投与後に生じた副作用のうちもっとも頻度が高かったのは発熱で、入院期間の延長が必要となった副作用も投与例数16名のうち2名の発熱のみで、安全性の高い治療であることが示されました。
ウイルス療法は、がん細胞に感染させたウイルスが増えることによって直接がん細胞を破壊する手法で、革新的ながん治療法として期待されます。G47Δの開発は、発明から医師主導治験に至るまで、研究者だけで推し進めてきました。本治験でG47Δの高い有効性が確認されたことを受けて、悪性神経膠腫(膠芽腫を含む悪性脳腫瘍の種類)を適応症としたG47Δの製造販売承認申請(注2)を行います。G47Δの開発は、世界に先駆けて日本で進められており、厚生労働省の先駆け審査指定制度(注3)および悪性神経膠腫を対象とした希少疾病用再生医療等製品(注4)の指定を受けています。承認されれば、日本で初めて実用化されるがん治療ウイルス薬となる見込みです。本治験における被験者登録はすでに終了しており、治験結果の詳細は、第11回日米癌合同会議(2019年2月12日17時:ハワイ時間/2019年2月13日12時:日本時間)において発表されました。
発表者
膠芽腫について
がんのウイルス療法とは
がんのウイルス療法とは、がん細胞のみで増えることができるウイルスを感染させ、ウイルスが直接がん細胞を破壊する治療法です。ウイルス療法では、遺伝子工学技術を用いてウイルスゲノムを「設計」して、がん細胞ではよく増えても正常細胞では全く増えないウイルスを人工的に造って臨床に応用します。がん細胞だけで増えるように工夫された遺伝子組換えウイルスは、がん細胞に感染するとすぐに増殖を開始し、その過程で感染したがん細胞を死滅させます。増殖したウイルスはさらに周囲に散らばって再びがん細胞に感染し、ウイルス増殖、細胞死、感染を繰り返してがん細胞を次々に破壊していきます。一方、正常細胞に感染した遺伝子組換えウイルスは増殖できないような仕組みを備えているため、正常組織は傷つきません(図1)。
1990年代以降に欧米で始まった、遺伝子組換えウイルスを用いたウイルス療法の臨床開発は、近年世界で競争が加速しています。中でも、単純ヘルペスウイルス1型を応用した開発が先頭を走っており、2015年10月には大手製薬企業が開発した第二世代のがん治療用ウイルス開発品(talimogene laherparepvec)が悪性黒色腫の治療薬として米国で認可され、続いて欧州でも認可されました。
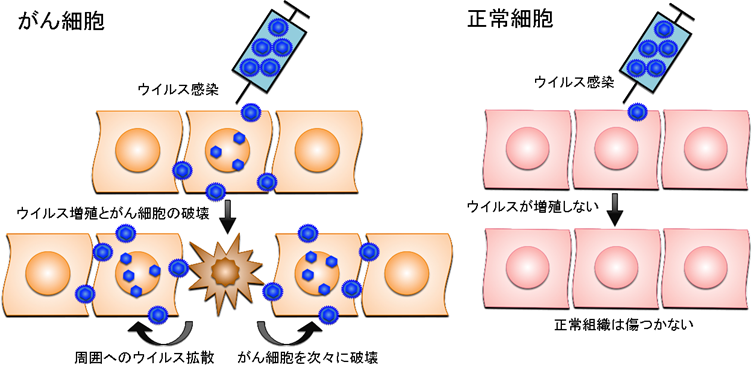
図1
G47Δ(ジーよんじゅうななデルタ)とは
G47Δは、口唇に水疱ができる口唇ヘルペスの原因ウイルスとして知られている単純ヘルペスウイルス1型の3つのウイルス遺伝子を改変して、藤堂教授らが作製した世界初の第三世代のがん治療用遺伝子組換えヘルペスウイルスです(図2)。
単純ヘルペスウイルス1型は、がん治療に有利な特長を多く備えています。その主な特長は、1)ヒトのあらゆる種類の細胞に感染できること、2)細胞を殺す力が比較的強いこと、3)抗ウイルス薬が存在するため治療を中断できること、4)患者がウイルスに対する抗体を持っていても治療効果が弱くならないこと、などです。単純ヘルペスウイルス1型のゲノムから、正常細胞での増殖では必要でがん細胞では不要なウイルス遺伝子を取り除くことで、がん細胞だけで増えるウイルスを造ることができます。
3つのウイルス遺伝子を改変したG47Δは、既存のがん治療用ウイルスに比べて安全性と治療効果が格段に高くなっています。また、G47Δの特徴としてがん細胞を破壊する過程で、抗腫瘍免疫を惹起するために、G47Δを投与した部位のみならず、投与していないところにあるがんにも免疫を介して効果が期待できます。さらに、G47Δは、がんの根治を阻むとされるがん幹細胞をも効率よく破壊することが判っています。
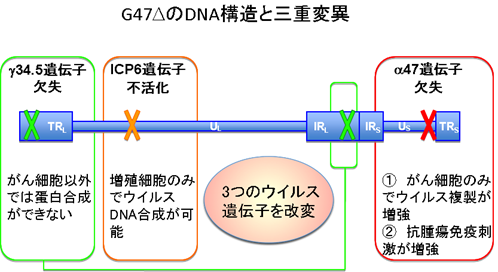
図2
医師主導治験
臨床試験は、欧米ではクリニカル・トライアル(clinical trial)と一つの言葉で表されますが、日本では臨床研究と治験という2種類に区別され、それぞれ異なる法令や審査ルートに従う必要があります。国から医薬品としての承認(製造販売承認)を受け、実臨床において患者が薬として使えるようにするためには、医薬品医療機器総合機構を窓口とする「治験」を行って臨床データを集めなければなりません。2003年に薬事法が改正され、製薬企業等と同様に治験の準備から管理を医師自ら行うことができるようになり、これを医師主導治験といいます。医師主導治験では医師自ら治験を実施できるようになりましたが、治験実施計画書等の作成から始まり、治験計画届の提出、治験の実施、モニタリングや監査の管理、試験結果を取りまとめた総括報告書の作成など、治験のすべての業務を医師自らが実施して統括しなければならず、多大な資金と労力を必要とします。
G47Δの臨床開発
G47Δは東京大学の藤堂教授らが開発した革新的ながん治療用ウイルスであり、世界に先駆けて日本で臨床開発を展開しています。G47Δの臨床開発は、真のアカデミア発のトランスレーショナルリサーチ(注5)として進められました。培養細胞や動物を用いた安全性や有効性の試験はもとより、臨床試験に用いる治験薬の製造も東京大学医科学研究所内の施設で研究チームが自ら行いました。G47Δを初めてヒトに投与するいわゆるファースト・イン・マン(first-in-man)臨床試験は、2009年から、膠芽腫を対象とした臨床研究として東京大学で5年間実施され、脳腫瘍内への投与が安全であることが確認されました。今回の治験は、有効性を検討する第Ⅱ相からスタートする日本初のウイルス療法の治験として、2015年5月に被験者登録を開始しました。この医師主導治験は、外国で承認されていながら国内未承認、あるいは適応外使用が一般的となっている医薬品や医療機器について実施するものとは異なり、非臨床試験から治験薬製造、規制対応、治験実施まで製薬企業が全く関与せずにアカデミアだけで行ったという点で、日本の医薬品開発の歴史に残るアカデミア主導創薬の成功例と言えます。今回の医師主導治験により、ウイルス療法薬としては日本で初めてG47Δの治療効果が確認されました。単純ヘルペスウイルス1型を用いたウイルス療法が悪性脳腫瘍で治療効果を示すことが臨床試験で確認されたのは、今回が世界で初めてです。
本治験の概要
- 対象疾患:
- 初期治療後に残存または再発した膠芽腫
- 試験デザイン:
- 第Ⅱ相(注1)。医師主導治験。対照群のないオープンラベル試験
- 実施施設:
- 単施設(東京大学医科学研究所附属病院)
- 投与方法:
- 定位脳手術による腫瘍内投与。最大6回までの繰り返し投与
1回目と2回目は5~14日の間隔、3回目以降は4週間の間隔で投与 - 予定された被験者数:
- 30名
- 主要評価項目:
- 一年生存割合(治療開始後1年間生存した患者の割合)
- 副次評価項目:
- 全生存期間、無増悪生存期間、腫瘍縮小効果
- 被験者登録期間:
- 2015年5月~2018年7月
- 観察期間:
- 治療終了後2年間
中間解析結果の概要
本治験は、初期治療後に残存または再発した膠芽腫病変を有する、膠芽腫の中でも予後が悪い患者を対象としました。最初の手術後に、放射線照射と化学療法(テモゾロミド)を行うという標準治療に対して、G47Δを用いたウイルス療法を上乗せした場合に、生存期間を延長させることを有効性として検討する治験デザインとなっています。30名の被験者を予定して2015年5月から被験者登録を開始しました。当初より、13名の被験者が治療開始から1年経過した時点で中間解析を実施する治験実施計画となっており、2018年7月に中間解析を実施しました。その結果、主要評価項目(G47Δの治療効果を判定する基準)に設定していた1年生存割合が92.3%(13例中12例が治療開始後1年以上生存)を示し、標準治療の1年生存割合15%(他の複数の臨床試験結果から算出)に比べ格段に高く、G47Δが高い治療効果を呈することが確認されました。一方、中間解析の時点で、16名の被験者を対象に安全性の解析を行いました。主な副作用は発熱が15名(93.8%)、嘔吐及びリンパ球数減少が各8名(50.0%)、悪心が7名(43.8%)でした。入院期間の延長が必要となった副作用は、発熱2名(いずれもGrade 2 [軽度])(12.5%)のみでした。
この中間解析の結果、30名の被験者登録を完了するまでもなく、中間解析の時点で、本治験におけるG47Δの治療効果の有効性が確実となったため、治験実施計画に従い、独立データモニタリング委員会の勧告を経て、被験者登録を終了しました。治験実施計画により、治療を受けた被験者は、治療終了後2年間観察されます。
有効性の解析
- 主要評価項目(13名)
- 1年生存割合:92.3% (12/13名)
- 副次評価項目(16名)
- 全生存期間中央値:中間解析時点では中央値に達していない。
- 無増悪生存期間中央値: 8.6カ月
- 腫瘍縮小効果:2年間の経過観察が終了した4名では、最良効果はいずれもSD(Stable Disease [安定])。
- 安全性の解析(16名)
- 主な副作用:発熱(93.8%)、嘔吐(50.0%)、リンパ球数減少(50.0%)、悪心(43.8%)
- Grade 3 [中等度]以上の副作用:リンパ球数減少(25.0%)
- Grade 4 [重度]の副作用:リンパ球数減少(12.5%)
- 入院期間の延長が必要となった副作用:発熱(Grade 2 [軽度])(12.5%)
製造販売承認申請の見込み
本治験の結果を基に、現在、悪性神経膠腫を適応症としたG47Δの製造販売承認申請の準備を行っており、申請時期は、3~4ヶ月後を見込んでいます。製造販売承認申請は、第一三共株式会社が行います。G47Δの製品製造は、デンカ生研株式会社が行っています。G47Δは、厚生労働省の先駆け審査指定品目に指定されていますので、先駆け総合評価相談による事前評価の充実かつ優先審査等により、製造販売承認申請から6ヶ月後の承認を目指した審査期間の短縮が見込まれています。
今後の展開
この治験結果を基に、今回は、悪性神経膠腫を適応症としてG47Δの製造販売承認申請を行いますが、G47Δはあらゆる固形がんに有効であることが動物実験で示されており、今後さらに、可及的速やかに全ての固形がんに適応を広げることを目指します。2013年からからは、前立腺癌と嗅神経芽細胞腫をそれぞれ対象とした臨床試験を実施しました。2018年からは、悪性胸膜中皮腫の患者の胸腔内にG47Δを投与する臨床試験も開始しています。
G47Δの臨床開発への支援
- 文部科学省「がんトランスレーショナル・リサーチ事業―革新的ながん治療法等の開発に向けた研究の推進―」(平成16(2004)年~平成20(2008)年度)
- 文部科学省「橋渡し研究支援推進プログラム」(平成19(2007)年~平成23(2011)年度)
- 文部科学省「橋渡し研究加速ネットワークプログラム」(平成24(2012)年度~平成28(2016)年度)
- 内閣府「最先端研究開発支援プログラム」(平成21(2009)年~平成25(2013)年度)
- 厚生労働科学研究費補助金「難病・がん等の疾患分野の医療の実用化研究事業(がん関係研究分野)」(平成24(2012)年度~平成26(2014)年度)
- 日本医療研究開発機構(AMED)「革新的がん医療実用化研究事業」(平成27(2015)年度~現在)
- 日本医療研究開発機構(AMED)「革新的医療技術創出拠点プロジェクト/橋渡し研究戦略的推進プログラム」(平成29(2017)年度~現在)
用語説明
- (注1)第Ⅱ相臨床試験
- 医薬品の開発は一般的に、安全性を調べる第Ⅰ相、比較的少数の被験者を対象にして治療効果を調べる第Ⅱ相、多数の被験者を対象に治療効果を検証する第Ⅲ相の3段階で臨床試験を行います。今回の医師主導治験は、第Ⅱ相試験です。がんを対象とする医薬品開発の場合は、一般的に、標準治療で効果が得られなかった被験者を対象に第Ⅰ相試験を行います。第Ⅱ相試験は、一般に、開発する医薬品の治療効果を、標準治療と比べます。本治験でも、G47Δを用いたウイルス療法を標準治療に上乗せして、標準治療の治療成績より良い結果が得られるかどうかを検討する治験デザインとなっています。第Ⅲ相試験を行うためには、多数の被験者の参加が必要となります。悪性神経膠腫のような希少ながんの場合は、第Ⅲ相試験を行うことが困難であるため、しばしば第Ⅱ相試験までで有効性を確認します。G47Δは、悪性神経膠腫を対象とした希少疾病用再生医療等製品に指定されています。
- (注2)製造販売承認申請
- 医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律(医薬品医療機器等法)に基づいて、医薬品等の製造および販売の承認を厚生労働省に申請することです。G47Δのようなウイルス療法薬は、平成26年11月25日に施行された医薬品医療機器等法では再生医療等製品に分類されます。いわゆる製薬企業が申請できます。医薬品医療機器総合機構により、企業としての責任体制の審査、製品の有効性・安全性等の審査、製品の生産方法・管理体制の審査など、さまざまな審査がなされます。
- (注3)先駆け審査指定制度
- 世界最先端の治療薬を日本の患者に最も早く提供することを目指し、一定の要件を満たす画期的な新薬等について、開発の比較的早期の段階から先駆け審査指定制度の対象品目に指定し、製造販売承認の相談・審査において優先的な取扱いの対象とする厚生労働省の制度です。原則として既承認薬と異なる作用機序を持ち、高い有効性が期待される医薬品が指定されます。指定には以下の4つ要件をすべて満たす必要があります。
- 治療薬の画期性
- 対象疾患の重篤性
- 対象疾患に係る極めて高い有効性
- 世界に先駆けて日本で早期開発・申請する意思
- (注4)希少疾病用再生医療等製品
- 希少疾病用再生医療等製品は、医薬品医療機器法第77条の2に基づき、対象患者数が5万人未満で本邦では十分にその研究開発が進んでいない状況にあり、医療上特にその必要性が高いものに対し、次の指定基準に合致するものを厚生労働大臣が指定するもので、希少疾病用医薬品の試験研究を促進するための特別の支援措置を講ずる制度です。
- 対象者数
再生医療等製品の対象者の数が、本邦において5万人未満であること。 - 医療上の必要性
重篤な疾病を対象とするとともに、代替する適切な治療法がなく、既存の医薬品・医療機器・再生医療等製品と比較して著しく高い有効性又は安全性が期待され、特に医療上の必要性が高いものであること。 - 開発の可能性
対象疾病に対して当該再生医療等製品を使用する理論的根拠があるとともに、その開発に係る計画が妥当であると認められること。
- 対象者数
- (注5)トランスレーショナルリサーチ
- 医療につながる基礎研究成果を臨床における実用化に橋渡しする開発研究を指します。知的財産権の確保、臨床に用いる製剤の製造・品質試験・安定性試験、動物を用いた安全性試験、臨床試験実施計画の作成、規制対応など、数多くのプロセスを経ます。また、多大な開発資金と多大の労力を必要とします。
参照URL
お問い合わせ先
取材に関するお問合せ先
東京大学医科学研究所事務部管理課
電話:03-6409-2018(直通)(平日午前9時~午後5時)
e-mail:t-soumu”AT”ims.u-tokyo.ac.jp
東京大学医科学研究所附属先端医療研究センター 先端がん治療分野(脳腫瘍外科)
担当:藤堂教授室
電話:03-6409-2142(直通)
AMED事業に関するお問い合わせ先
革新的がん医療実用化研究事業:
国立研究開発法人日本医療研究開発機構(AMED)
戦略推進部 がん研究課
電話:03-6870-2221
e-mail:cancer”AT”amed.go.jp
革新的医療技術創出拠点プロジェクト/橋渡し研究戦略的推進プログラム:
国立研究開発法人日本医療研究開発機構(AMED)
臨床研究・治験基盤事業部 臨床研究課
電話:03-6870-2229
e-mail:rinsho”AT”amed.go.jp
※e-mailは上記アドレス"AT"の部分を@に変えてください。
掲載日 平成31年2月13日
最終更新日 平成31年2月13日