


脳内の免疫担当細胞ミクログリアのM1/M2極性転換分子スイッチを発見―リソソーム酵素カテプシン群の新たな機能を解明―
プレスリリース
概要
九州大学大学院歯学研究院の中西 博教授らの研究グループは、リソソーム酵素カテプシンEならびにカテプシンB(※1)によるプロテアーゼ反応のリレーが脳常在性ミクログリア(※2)のもつ傷害性(M1型)ならびに保護性(M2型)の極性転換における分子スイッチとして働くことをマウスの低酸素/脳虚血モデル(※3)を用いて明らかにしました。この研究成果は、脳炎症の慢性化ならびに神経傷害の発生メカニズムを理解するうえでの新たな知見をもたらし、カテプシンEならびにカテプシンBを標的とした新しい炎症性脳病態に対する治療薬開発の可能性を提示するものです。
なお、本研究は、国立研究開発法人日本医療研究開発機構 革新的先端研究開発支援事業(AMED-CREST)ならびに国立研究開発法人科学技術振興機構 戦略的創造研究推進事業(CREST)の一環として行われ、本研究成果は、2015年9月9日(水)(米国東部時間)に米国神経科学会誌『Journal of Neuroscience』にオンライン掲載されました。
背景
高齢化が急速に進行する日本において慢性炎症が原因となる疾患が急増しており、炎症の慢性化メカニズムの解明ならびに制御法の開発は急務となっています。炎症時に見られる浸潤性マクロファージは機能的に相反する傷害性(炎症性)に働くM1型ならびに保護性(抗炎症性)に働くM2型に分類され、同様に脳常在性ミクログリアについてもM1/M2分類が適用されるようになっています(図1)。これまでの研究では、このようなミクログリアの性質が傷害性、保護性に変化する「極性転換」において、炎症反応に必須な転写因子(NF-κB)(※4)の活性化が重要な役割を果たすことが知られていますが、どのような仕組みでミクログリアの性質が変化するのか、極性転換の「分子スイッチ」の全容は、未だ明らかになっていません。
本研究グループは、長年に渡り、ミクログリアの産生するリソソーム酵素カテプシン群のプロテアーゼ作用に着目し、これまでカテプシンが疼痛の発症や慢性化において重要な役割を果たしていることを明らかにしてきました。今回の研究では、カテプシン群がミクログリアの性質の変化にどのように関わっているのか、転写因子NF-κBの活性化への関連を調べ、カテプシン群がミクログリアの M1/M2極性転換における「分子スイッチ」として働く可能性について検討を行いました。
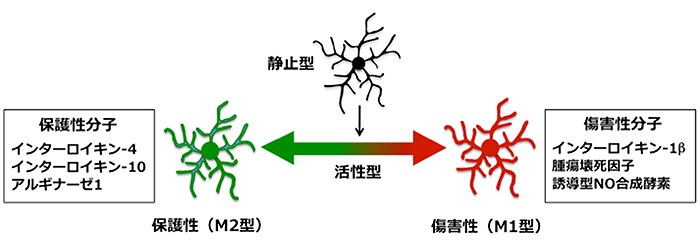 (図1)ミクログリアの相反する二つの極性:「傷害性(M1型)」ならびに「保護性(M2型)」
(図1)ミクログリアの相反する二つの極性:「傷害性(M1型)」ならびに「保護性(M2型)」
内容
脳の炎症状態モデルとして低酸素/脳虚血の負荷をかけた野性型マウスでは、海馬(※5)に重度の神経傷害が生じていること、神経傷害に先行して海馬に集積したミクログリアにカテプシンBが増大していることがわかりました。一方、カテプシンBの機能を明らかにする目的で、カテプシンBを欠損させたマウスを作成したところ、神経傷害が有意に軽減していることがわかりました。低酸素/脳虚血を負荷した野性型マウスの海馬より経時的に単離したミクログリアでは、図1に示すような傷害性分子の持続的発現とともに、保護性分子の一過性発現が認められました。ところが、カテプシンBを欠損させたマウスの海馬より単離したミクログリアでは、保護性分子の早期からの一過性発現のみが認められました。さらに、培養系における脳虚血モデルである低酸素/低グルコースを負荷すると、培養ミクログリアにおいてオートファジー(※6)が誘導され、オートファジーの実行因子であるカテプシンBは、普段は転写因子NF-κBに結合することで不活性化させている抑制因子IκBα(※7)を分解し、転写因子NF-κBの核内移行を促進することで傷害性分子の遺伝子転写を誘導しました(図2: オートファジー経路)。
一方、カテプシンBと同様に、低酸素/脳虚血の負荷をかけた野生型マウスではカテプシンEが神経傷害に先行して海馬に集積したミクログリアで増大し、カテプシンEを欠損させたマウスでは神経障害が有意に軽減していました。興味深いことに、カテプシンEを欠損させたマウスでは、低酸素/脳虚血を負荷しても、ミクログリアにおけるカテプシンB増大は誘導されませんでした。また、低酸素/脳虚血を負荷した野性型マウスの海馬より経時的に単離したミクログリアでは、カテプシンEならびにTNF関連アポトーシス誘導リガンド(TRAIL)(※8)の発現が増大しました。低酸素/低グルコースを負荷した培養ミクログリアにおいて、培養上清にはカテプシンE に依存したTRAILの分泌が認められました。さらにTRAILの添加により、タンパク質分解酵素複合体のプロテアソームは修飾タンパク質であるユビキチンを目印として抑制因子IκBαを分解し、転写因子NF-κBの活性化ならびにカテプシンBの発現増大を誘導しました(図2: プロテアソーム経路)。
以上の結果より、次のことがわかりました。低酸素/脳虚血に伴い、ミクログリアで増大したカテプシンEは、ミクログリアの細胞膜に結合して存在するTRAIL(膜結合型)を遊離します。遊離したTRAIL(可溶型)は、ミクログリアに発現するTRAIL受容体を介しプロテアソームによる抑制因子IκBαの分解を促進し、転写因子NF-κBの活性化を急性に引き起こします。さらに、転写因子NF-κBはオートファジーの誘導ならびにカテプシンBの発現増大を引き起こし、オートファジーの実行因子であるカテプシンBによる抑制因子IκBαの分解をさらに誘導します。これらの2つのプロテアーゼ反応のリレーによって、最終的に転写因子NF-κBの慢性的活性化を引き起こし、神経傷害性の性質を持ったミクログリア(M1型)に変化させることが明らかとなりました(図2)。
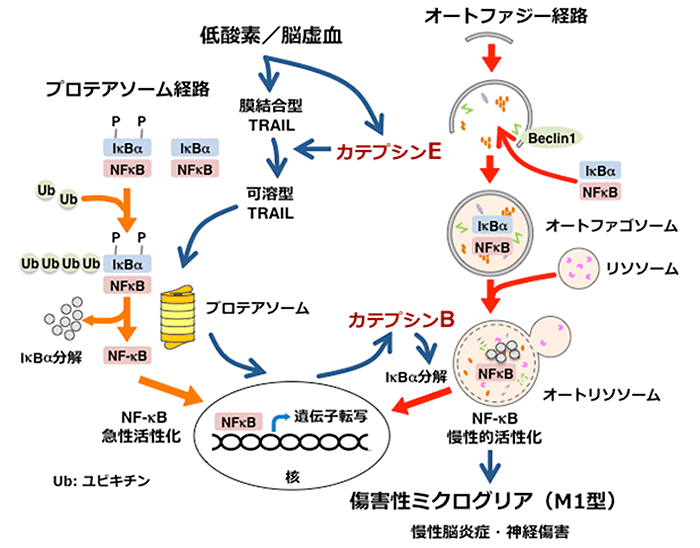 (図2)低酸素/脳虚血におけるミクログリアの傷害性変化の細胞内プロセス
(図2)低酸素/脳虚血におけるミクログリアの傷害性変化の細胞内プロセス
効果・今後の展開
また、神経傷害性に変化したミクログリア(M1型)は、慢性脳炎症ならびに神経傷害を誘導すると考えられています。そこで今後は、末梢に投与可能なカテプシンEならびにカテプシンBの特異的阻害剤を創出し、炎症性脳病態に対する治療薬としての有効性について検討を行う予定です。
用語解説
- (※1)カテプシンEならびにカテプシンB:
- 両者ともリソソーム性プロテアーゼの一種
- (※2)ミクログリア:
- 脳脊髄に存在し免疫機能を担う中枢神経中のグリア細胞の一種
- (※3)低酸素/虚血モデル:
- 片側総頸動脈閉塞による脳虚血を行った後、酸素濃度8%環境による低酸素を負荷する脳虚血モデル
- (※4)転写因子NF-κB:
- 主に炎症性分子の転写因子として働くタンパク質複合体
- (※5)海馬:
- 記憶や空間学習能力に関わる脳部位
- (※6)オートファジー:
- 細胞が持っている細胞内のタンパク質を分解するための仕組みの一つ
- (※7)抑制因子IκBα:
- 転写因子NF-κBに結合子し核移行シグナルを阻害することで不活性化するタンパク質で、抑制因子IκBα の分解により転写因子NF-κBは活性化される
- (※8)TNF関連アポトーシス誘導リガンド(TRAIL):
- 受容体であるDR5に結合し、アポトーシスや転写因子NF-κBの活性化を誘導するTNFスーパーファミリーに属するリガンド
本研究について
お問い合わせ先
研究に関するお問い合わせ
九州大学大学院歯学研究院 教授
中西 博 (なかにし ひろし)
電話:092-642-6413 FAX:092-642-6215
E-mail:nakan“at”dent.kyushu-u.ac.jp
革新的先端開発支援事業に関するお問い合わせ
国立研究開発法人日本医療研究開発機構(AMED)
戦略推進部 研究企画課
〒100-0004 東京都千代田区大手町一丁目7番1号
TEL:03-6870-2224
E-mail:kenkyuk-ask“at”amed.go.jp
※E-mailは上記アドレス“at”の部分を@に変えてください。
関連リンク
掲載日 平成27年9月10日
最終更新日 平成27年9月10日