


筋萎縮性側索硬化症(ALS)発症の仕組みの一端を解明
プレスリリース
国立研究開発法人日本医療研究開発機構
概要
大阪市立大学大学院医学研究科 分子病態学の徳永文稔(とくなが ふみのり)教授らの研究グループは、東京大学大学院理学系研究科 濡木理(ぬれき おさむ)教授、和歌山県立医科大学神経内科 伊東秀文(いとう ひでふみ)教授らの研究チームとともに、家族性筋萎縮性側索硬化症(きんいしゅくせいそくさくこうかしょう)(ALS)の原因遺伝子であるオプチニューリン(optineurin)の研究を行い、ALS発症メカニズムの一端を明らかにしました。
ALSは、運動神経細胞(ニューロン)が選択的に侵される神経難病で、多くは意識がはっきりしたまま、筋力低下のため歩行困難や構音障害(発音が正しくできない症状)となり、呼吸不全に至りますが、根本的な治療法はありません。ALSのおよそ90%は、発症原因が不明な孤発性ALSですが、約10%は遺伝子変異が関連する家族性ALSです。これまでに約20の原因遺伝子が見出され、その解析からALS発症機構解明を目指す研究が進められています。オプチニューリンの遺伝子変異がALSに関わることは2010年に日本で発見され、重要な病因因子であることが明らかにされています。
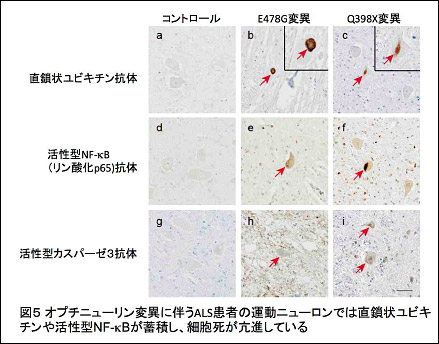
以前、私たちはユビキチンという低分子たんぱく質が特異的な連結をした「直鎖状ユビキチン鎖」を発見し、この特異的構造体がたんぱく質分解ではなく、炎症や免疫に重要なNF-κB(エヌエフ-カッパービー)を介したシグナル伝達経路を活性化することを見出していました。そして今回の研究で、オプチニューリンが直鎖状ユビキチン鎖に選択的に結合し、NF-κBや細胞死を抑制していることを突き止め、実際にオプチニューリン変異に起因するALS患者の運動ニューロンでは、直鎖状ユビキチン鎖や活性化 NF-κBが蓄積し、神経細胞死を引き起こしていることを明らかにしました。本研究から直鎖状ユビキチン鎖生成を介する慢性的な神経炎症の亢進が神経細胞死を引き起こすことが明らかになり、今後、ALS治療の標的になる可能性が示唆されました。本研究の成果は、平成28年8月24日(水)午前10時(英国現地時間)、日本時間では平成28年8月24日(水)午後6時に英国の科学雑誌ネイチャー・コミュニケーションズにオンライン掲載されます。
- 【発表雑誌】
- Nature Communications
- 【論文名】
- Linear ubiquitination is involved in the pathogenesis of optineurin-associated amyotrophic lateral sclerosis
「オプチニューリン異常に伴う筋萎縮性側索硬化症の病因に直鎖状ユビキチン化が関与する」 - 【著者】
- Seshiru Nakazawa, Daisuke Oikawa, Ryohei Ishii, Takashi Ayaki, Hirotaka Takahashi, Hiroyuki Takeda, Ryuichiro Ishitani, Kiyoko Kamei, Izumi Takeyoshi, Hideshi Kawakami, Kazuhiro Iwai, Izuho Hatada, Tatsuya Sawasaki, Hidefumi Ito, Osamu Nureki, and Fuminori Tokunaga
- 【掲載URL】
- http://www.nature.com/ncomms/index.html
研究の背景
ユビキチンは、細胞内の不要たんぱく質を分解に導く標識因子として見出されましたが、近年ではたんぱく質分解のみならず、ユビキチンの連結様式の多様性によって多彩な生理機能を調節することが明らかになっています(用語解説参照)。
これまで知られているユビキチンの連結は、いわばジグサグ状(分子内のリシンというアミノ酸を介したイソペプチド結合による分岐鎖状ポリユビキチン鎖)でしたが、私たちは真っ直ぐなタイプのユビキチン連結鎖(直鎖状ユビキチン鎖)を作ることができる酵素LUBAC(ルーバック)を世界に先駆けて見出しました。LUBACは直鎖状ユビキチン鎖を生成できる唯一の酵素です。さらに私たちは、LUBACによる直鎖状ユビキチン鎖生成は、炎症や免疫応答の制御に重要なNF-κBシグナル活性化の足場として機能することを明らかにしました(2009年、Nature Cell Biology誌に発表)(図1)。さらに、LUBACの生理的な新規サブユニットとしてSharpinを同定し、Sharpinが欠損したマウスでは慢性増殖性皮膚炎を引き起こすことを見出しました(2011年、Nature誌に発表)。また、LUBACによる直鎖状ユビキチン鎖生成を介したNF-κB活性化に拮抗する脱ユビキチン化酵素(A20)の解析から、A20は直鎖状ユビキチン鎖に結合する領域を持っており、これがNF-κB活性制御に重要であることや、その変異はB細胞リンパ腫発症に関わることを明らかにしました(2012年、EMBOJ誌に発表)。現在、直鎖状ユビキチン鎖を中心とした新しい炎症・免疫制御機構と疾患との関連の研究が世界的に高く注目されています。
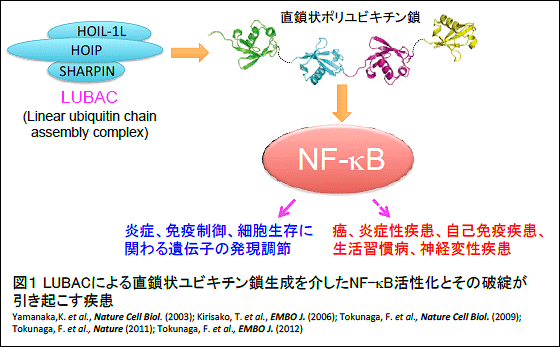
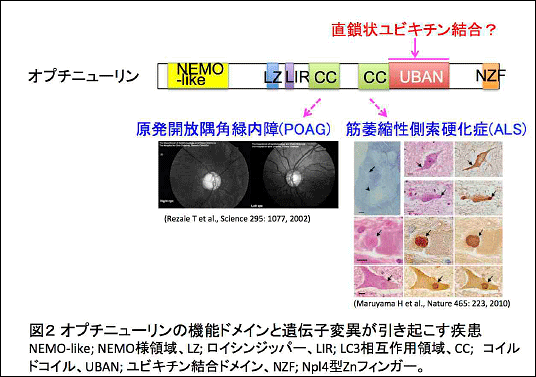
研究の内容
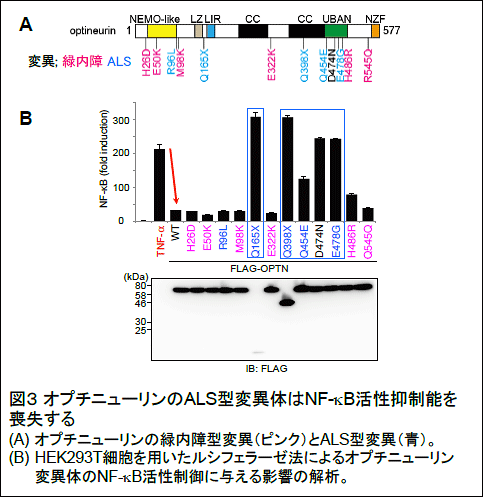
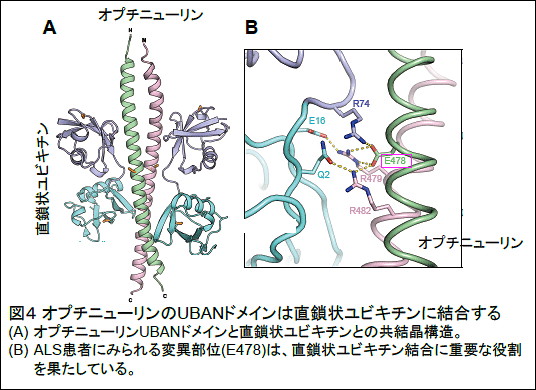
これらの結果から、オプチニューリンは生理的にはNF-κB活性やアポトーシスを抑制し、細胞の生死に関わる重要なシグナル伝達経路を制御することが示されました(図6左)。一方、ALSを引き起こすオプチニューリン変異では、直鎖状ユビキチン鎖に結合できないためNF-κB活性が恒常的に亢進すると考えられます。このため、患者の運動ニューロンでは、直鎖状ユビキチンや活性型 NF-κB因子(P-p65)がサイトゾル凝集体に蓄積し、細胞死(アポトーシス反応)も亢進すると示唆されました(図6右)。この様に、本研究からオプチニューリンの機能不全によって神経炎症の持続的亢進と神経細胞死の促進がおこり、これがALS発症に関わる可能性が示されました。
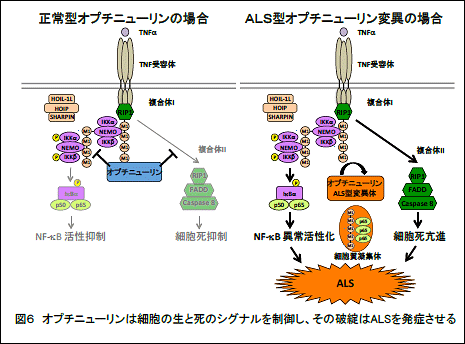
本研究の意義
ALSの発症機構としては、ダメージを受けたたんぱく質やRNAとたんぱく質との複合体などが構造異常となり、たんぱく質分解機構(プロテアソーム系やオートファジー系)で適切に分解されず細胞内に蓄積すること、これがNF-κBシグナルの活性化を介して神経炎症を引き起こすことが重要と考えられています(図7)。これまでも各種神経変性疾患でユビキチン陽性凝集体が細胞内に蓄積していることはよく知られており、これはたんぱく質分解の不全に由来すると考えられていましたが、今回の研究からたんぱく質分解ではなく、炎症惹起に関わる「直鎖状ユビキチン鎖」がサイトゾル凝集体に局在しており、神経細胞死に関わるという全く新しい知見を得ました。本研究は、ALS発症の新たな細胞機構を示したものといえます。
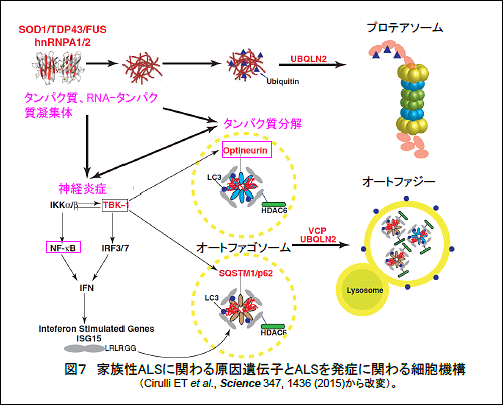
期待される効果
本研究はLUBACによる直鎖状ユビキチン鎖生成を介したNF-κB活性化や、その制御破綻がALS発症に関わる可能性を示したもので、今後、LUBAC活性阻害剤がALS治療薬開発の標的となるなど、新たな研究進展が期待されます。
今後の展開について
本研究ではオプチニューリン異常に由来するALSに直鎖状ユビキチン鎖の生成・蓄積が関わることが分かりましたが、今後、孤発性ALSや他の遺伝子異常に由来するALSなど研究の範囲を広げることで、本研究で見出されたメカニズムの重要性を解析することが必要です。さらに、アルツハイマー病やパーキンソン病などユビキチン陽性凝集体形成を伴う各種神経変性疾患において、どのような連結様式のユビキチン鎖が蓄積しているか解析することで疾患発症を引き起こす細胞機構解明の新たな展開が期待されます。また、ユビキチン修飾系やNF-κBシグナル伝達経路因子を標的とした機能抑制性化合物が新たな創薬へ繋がる可能性があります。この様な基礎研究からALSという難病発症の仕組みの一端が解明され、根本的治療法確立へ一歩を進めることができるよう、今後も研究を進めて参ります。
本研究は、国立研究開発法人日本医療研究開発機構(AMED)の革新的先端研究開発支援事業(AMED-CREST)「炎症の慢性化機構の解明と制御に向けた基盤技術の創出」研究開発領域(研究開発総括:宮坂昌之)における研究開発課題「慢性炎症による疾患発症機構の構造基盤」(研究開発代表者:濡木理)の一環で行われました。なお、本研究開発領域は、平成27年4月の日本医療研究開発機構の発足に伴い、国立研究開発法人科学技術振興機構(JST)より移管されたものです。また一部、科研費等の支援も受けています。
用語解説
- ◎ユビキチン
- ユビキチンは、酵母からヒトまでの真核生物で高度に保存された低分子量たんぱく質で、細胞内で不要になったたんぱく質に結合することで、分解へ導く標識としてはたらく。このシステムを同定したイスラエルの研究者(アブラム・ハーシュコ、アーロン・チカノバー、アーヴィン・ローズ)らは2004年にノーベル化学賞を受賞した。その後の研究から、ユビキチンは多様な連結をすることで、たんぱく質分解だけでなく、DNA修復やシグナル伝達、細胞内膜輸送など多彩な役割を果たすことが明らかになっている。
- ◎NF-κB
- NF-κBは、1986年に米国のデイヴィッド・バルチモアーによって見出されたたんぱく質で、炎症や免疫制御に関わる500種以上の遺伝子の発現調節を司る転写因子である。NF-κB活性化経路の破綻は、多くの癌、炎症性疾患(炎症性大腸炎など)、自己免疫疾患(関節リウマチなど)、生活習慣病(糖尿病、肥満など)、神経変性疾患(アルツハイマー病、パーキンソン病、ALSなど)に関わることから、創薬標的として臨床的にも高く注目されている。
お問い合わせ先
研究内容に関するお問い合わせ先
大阪市立大学大学院 医学研究科
分子病態学 教授 徳永 文稔
TEL:06-6645-3720
FAX:06-6645-3721
E-mail:ftokunaga“AT”med.osaka-cu.ac.jp
報道に関するお問合せ先
大阪市立大学法人運営本部 広報室
担当:竹谷
TEL:06-6605-3411
FAX:06-6605-3572
E-mail:t-koho“AT”ado.osaka-cu.ac.jp
AMED 事業に関するお問い合わせ先
国立研究開発法人 日本医療研究開発機構(AMED)
戦略推進部 研究企画課
TEL:03-6870-2224
FAX:03-6870-2243
E-mail:kenkyuk-ask“AT”amed.go.jp
※E-mailは上記アドレス“AT”の部分を@に変えてください。
掲載日 平成28年8月24日
最終更新日 平成28年8月24日