プレスリリース
国立大学法人東京医科歯科大学
国立大学法人山梨大学
京都府立医科大学
国立研究開発法人日本医療研究開発機構
ポイント
- 上皮系マーカー遺伝子であるCDH1(E-カドヘリン)を用いたレポーターシステムと1,090種類のマイクロRNAを搭載したマイクロRNAライブラリーを組み合わせて上皮間葉転換(EMT)に関わる2種類のマイクロRNA(miR-509-5pとmiR-1243)を同定しました。
- miR-509-5pとmiR-1243は、それぞれ異なる経路でEMTを抑制することがわかりました。
- これらマイクロRNAを介して、EMTを抑制することで、膵がん治療で最も使用される抗がん剤のゲムシタビンの効果が上昇することがわかりました。
- 上記マイクロRNAの発現を調べることにより、ゲムシタビンの効果を予測できる可能性があります。
東京医科歯科大学・難治疾患研究所・分子細胞遺伝分野の村松智輝助教、稲澤譲治教授と京都府立医科大学大学院医学研究科消化器外科学 平本秀一大学院生、大辻英吾教授、ならびに、山梨大学医学部外科学講座第一教室 市川大輔教授らの研究グループは、独自のEMT可視化システムとマイクロRNAライブラリーを組み合わせることにより、上皮間葉転換(EMT)を制御するマイクロRNA(miRNA)を同定しました。この研究は、文部科学省新学術領域研究(15H05908)「がんシステムの新次元俯瞰と攻略」、国立研究開発法人日本医療研究開発機構(AMED)「次世代がん医療創生研究事業」(P-CREATE)などの支援のもと遂行され、その研究成果は、国際科学雑誌 Scientific Reports(サイエンティフィック リポーツ)に、2017年6月21日にオンライン版で発表されました。
研究の背景
がん転移は、予後不良の最たる原因であり、複雑かつ多段階のステップによって成立することが今までの研究から明らかになってきています。しかし、未だ不明な点が多く残されており、その分子機構の解明はがん治療薬開発における喫緊の課題です。がん転移のステップの一部としてよく知られているのが、上皮間葉転換(EMT: Epithelial Mesenchymal Transition)です。EMTは、上皮系の細胞が、間葉系形質を獲得する現象であり、間葉系形質を獲得したがん細胞は移動・浸潤能が亢進し、がん転移を起こしやすいと考えられています。EMTを促進する遺伝子やEMTとは逆方向の間葉系形質から上皮系形質へ誘導する(MET: Mesenchymal Epithelial Transition)遺伝子も、数多く報告されており、その分子機構は徐々に解明されてきています。本研究では、独自に開発したレポーターシステム(EMT可視化システム [図1 A])と1090種類のマイクロRNA(miRNA)を搭載したmiRNAライブラリーを組み合わせた大規模EMT関連miRNAスクリーニングを用いて、EMT抑制性のmiRNAを同定することを目的として解析を行いました。このレポーターシステムは、上皮系マーカー遺伝子であるCDH1(E-カドヘリン)のプロモーター領域を蛍光タンパク質であるGFP(Green Fluorescent Protein)遺伝子の上流に挿入することにより、CDH1の転写活性依存的にGFPが発現するものです。このシステムを用いることにより、EMT変化をGFPの強度により可視化・定量化することが可能です。一方、miRNAとは、20~25塩基程度のRNAであり、複数の標的遺伝子を持ち、標的遺伝子の3’UTR(3’untranslated region)に結合し、翻訳または転写を抑制することが知られています。以前、研究グループでは、同EMT可視化システムと470種類のmiRNAを搭載したmiRNAライブラリーを用いてEMT抑制性のmiR-655を同定しており(Harazono Y et al., PLoS One 2013)、本研究ではさらに多くのmiRNAを対象に大規模スクリーニングを行いました。
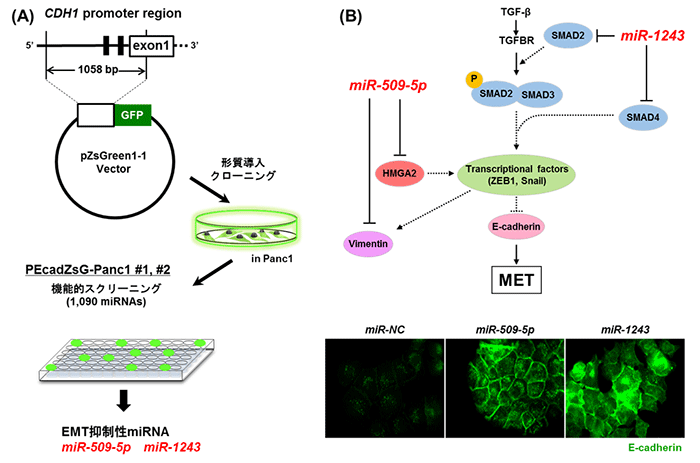 図1(A)EMT可視化システムの概要 (B)miR-509-5pとmiR-1243のEMT抑制シグナル経路
図1(A)EMT可視化システムの概要 (B)miR-509-5pとmiR-1243のEMT抑制シグナル経路
研究成果の概要
研究グループは、膵臓がん細胞株(Panc1)を用いてEMT可視化システムを構築し、1090種類のmiRNAをそれぞれ形質導入した後、GFPの蛍光強度を測定しました。また、EMT可視化システムの構築に際しては、スクリーニング精度を向上させるため、2つの独立した細胞を樹立しました。miRNA導入後、細胞増殖を著しく阻害するmiRNAは、候補miRNAから除外しました。その結果、2つの細胞株から共通して得られたEMT抑制性候補miRNAは、6つに絞られました。その後、検証を重ね、最終的にEMT抑制性miRNAとしてmiR-509-5pとmiR-1243を同定しました。miR-509-5pは、EMT促進性遺伝子として知られているHMGA2や間葉系マーカー遺伝子であるvimentinを直接翻訳抑制することによってMETを誘導します。一方、miR-1243は、EMT誘導経路の代表と知られるTGF-β経路の重要な構成因子であるSMAD2, 4を直接的に翻訳抑制することにより、METを引き起こします(図1 B)。次に、EMTを起こした細胞は、抗がん剤に耐性を獲得することが知られているため、研究グループはこれらmiRNAを細胞に導入した際に抗がん剤(ゲムシタビン)の効果が上昇するかどうかの検討を行いました。その結果、それぞれのmiRNAを導入後、ゲムシタビンの抗腫瘍効果を測定したところ、コントロール群に比べ、miRNA導入群ではゲムシタビンの効果が増強されることがわかりました(図2 A)。このことから、この2つのmiRNAは、METを促進させることにより、抗がん剤の感受性を高める効果があると考えられます。また、ヒト膵臓がん組織中におけるmiR-509-5pの発現を検討したところ、発現の低い群に比べ発現の高い群では予後が良好であるということがわかりました(図2 B)。
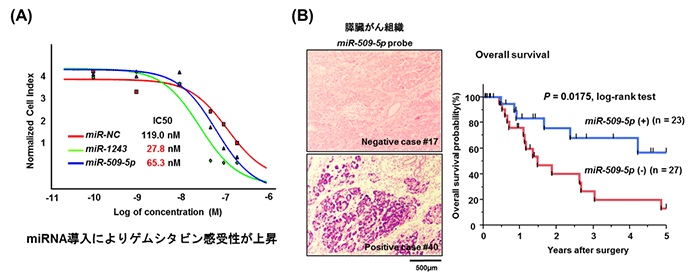 図2(A)miR-509-5pおよびmiR-1243導入によるゲムシタビン感受性の検討
図2(A)miR-509-5pおよびmiR-1243導入によるゲムシタビン感受性の検討
(B)膵臓がん組織中のmiR-509-5p発現(左)と生存曲線(右)
研究成果の意義
本研究では、EMT可視化システムとmiRNAライブラリーを組み合わせた大規模スクリーニングによってEMT抑制性miRNAであるmiR-509-5pとmiR-1243を同定することができました。このEMT可視化システムは、他の機能的ライブラリー(cDNA、siRNA、shRNA、化合物など)にも応用可能であり、EMT分子機構の解明にとって有益なツールです。本研究で同定したmiR-509-5pとmiR-1243のがん組織中での発現を調べることにより、予後のバイオマーカーとしてだけではなく、抗がん剤の効果を予測することができる可能性があります。また、上記miRNAを対象とした核酸薬の開発が成されれば、がん組織中において上記miRNAの発現がない場合においても、抗がん剤との併用治療を行うことにより、腫瘍抑制効果を増強できる可能性があります。
問い合わせ先
研究に関すること
東京医科歯科大学大学 難治疾患研究所
分子細胞遺伝分野
稲澤 譲治(イナザワ ジョウジ)
村松 智輝(ムラマツ トモキ)
TEL:03-5803-5820 FAX:03-5803-0244
E-mail:johinaz.cgen“AT”mri.tmd.ac.jp
京都府立医科大学大学院 医学研究科
消化器外科学
大辻 英吾(オオツジ エイゴ)
平本 秀一(ヒラモト ヒデカズ)
TEL:075-251-5527 FAX:075-251-5522
E-mail:otsuji“AT”koto.kpu-m.ac.jp
山梨大学 医学部外科学講座 第1教室
市川 大輔(イチカワ ダイスケ)
TEL・FAX:055-273-7390
E-mail:dichikawa“AT”yamanashi.ac.jp
AMED事業に関すること
国立研究開発法人日本医療研究開発機構(AMED)
戦略推進部 がん研究課
次世代がん医療創生研究事業担当
〒100-0004 東京都千代田区大手町1-7-1
TEL:03-6870-2221 FAX:03-6870-2244
E-mail:cancer“AT”amed.go.jp
報道に関すること
東京医科歯科大学 総務部総務秘書課広報係
〒113-8510 東京都文京区湯島1-5-45
TEL:03-5803-5833 FAX:03-5803-0272
E-mail:kouhou.adm“AT”tmd.ac.jp
京都府立医科大学 広報センター
[事務局:研究支援課]担当 中尾
TEL・FAX:075-251-5275
E-mail:kouhou“AT”koto.kpu-m.ac.jp
山梨大学 総務部総務課広報企画室
〒400-8510 山梨県甲府市武田4-4-37
TEL:055-220-8006 FAX:055-220-8799
E-mail:koho“AT”yamanashi.ac.jp
※Emailは上記アドレス“AT”の部分を@に変えてください。


