


「日本人基準ゲノム配列」初版JG1の公開―日本人のゲノム解析がこれまでよりも精密かつ正確に―
プレスリリース
東北大学東北メディカル・メガバンク機構
東北大学未来型医療創成センター
日本医療研究開発機構
発表のポイント
- 東北大学東北メディカル・メガバンク機構(ToMMo)は、日本人のゲノム解析を行うためのひな型となる基準ゲノム配列として「日本人基準ゲノム配列(JRGA)」の初版 JG1 を作成・公開しました。
- ゲノム医療の推進のためには、正確な個人のゲノム配列の解析が重要で、現在は、「基準となるゲノム配列」に対して、調べたい個人との差を検出する方法がよく用いられています。現在一般的に用いられている「基準となるゲノム配列」は、ヨーロッパ系とアフリカ系の集団を祖先に持つゲノムをもとに作成されたもので、「国際基準ゲノム*1」と呼ばれています。しかし、解析対象が日本人の場合には解析が困難な箇所(難読領域)があり、本来あるべき違いが検出されなかったり、誤検出があったりするなどの問題点が指摘されていました。
- 今回、ToMMoは、日本人3名に由来するゲノム配列を、複数の方法を組み合わせて、世界で初めて精密に決定し、JG1を作成しました。JG1をもとに、日本人のゲノム解析が行われることで、難読領域の解析をはじめ、解析の精度が向上することが見込まれます。JG1は研究や臨床のゲノム解析に広く利用できるようにインターネット上で公開します。
概要
現在ヒトの全ゲノム解析において主流となっている解析方法は、“短鎖技術による次世代シークエンシング解析”と呼ばれるもので、ゲノムを数百塩基程度までの短い単位(短鎖)で読み取り、基準となるゲノム配列に当てはめて配列を決めていきます。また、この当てはめを行う際のひな型として、国際基準ゲノム配列が用いられています。この方法は、科学研究を目的としたゲノム解析だけでなく、医療においてがんや遺伝性疾患の原因究明を目的としたゲノム解析にも採用されているものであり、これまでにToMMoが実施した数千名の全ゲノム解析*2でもこの方法を用いました。
しかしながら、国際基準ゲノム配列は、ヨーロッパ系とアフリカ系の集団を祖先に持つゲノムをもとに作成されたものであり、これを日本人の全ゲノム解析のひな型として用いた場合には、ゲノムの民族集団差が十分に考慮できず、解析が困難な箇所(難読領域)が発生したり、本来あるべき違いが検出されなかったり、誤検出があったりするなどの問題点が指摘されていました。また、日本人では、遺伝性の原因が強く疑われる疾患に対しても半数程度の症例でしか原因となる遺伝子が同定できていませんが、これも全ゲノム解析の精度が十分ではないことが要因の一つと考えられます。
このような課題を克服するため、ToMMoでは、日本人に特有のゲノム領域も評価できるようにすべく、難読領域を含めた日本人の全ゲノム配列の解読に挑戦してきました。今回、ToMMoは、日本人3名に由来するゲノム配列を、複数の方法を組み合わせて、世界で初めて精密に決定し、日本人基準ゲノム配列の初版となるJG1を構築することに成功しました。
日本人の全ゲノム解析を行う際、国際基準ゲノム配列の代わりにJG1をひな型として用いることにより、日本人の全ゲノム解析をこれまでより高精度に行うことが可能になります。これにより、希少疾患の遺伝要因の究明、ToMMoが構築してきた日本人全ゲノムリファレンスパネルの精度向上、がんゲノム解析、さらには日本人特有の疾患感受性や特有の薬剤感受性に寄与するゲノム配列変化の解明などが大きく進展することが期待されます。
今回完成したJG1は、研究や臨床目的で広く利活用できるように公開いたしました。本研究は、文部科学省・日本医療研究開発機構(AMED)による東北メディカル・メガバンク計画(【参考】を参照)のもと東北大学東北メディカル・メガバンク機構によって行われ、東北大学未来型医療創成センター(INGEM)所属の研究者も参画しています。
詳細
国際基準ゲノムの問題点と日本人基準ゲノム配列の必要性
これまで全ゲノム解析に広く利用されてきた国際基準ゲノム配列は、複数の個人に由来するとされているが、実際は、配列の70%以上がヨーロッパ系とアフリカ系が同程度に混交している祖先をもつ単一の個人に由来している。そのため、次世代シークエンシング解析(次世代シークエンサーを用いた全ゲノム解析など)での利用において、次のような課題を含んでいる。まず、国際基準ゲノム配列には、単一の個人に由来する多くの希少なバリエーションが含まれてしまっていることにより、日本人での配列バリエーション検出に見落としや誤検出が生じる可能性である(レアリファレンスアレル問題)。また、国際基準ゲノム配列は、日本人から遠い祖先性のため、民族集団固有の構造バリアント*3などに起因した難読領域が存在する可能性がある。これらの問題を解決し、日本人での次世代シークエンシング解析を高精度化するためには、日本人に祖先性を持つ複数の全ゲノム配列を解読し、それを統合した日本人基準ゲノムを構築することが必要であった。(図1)
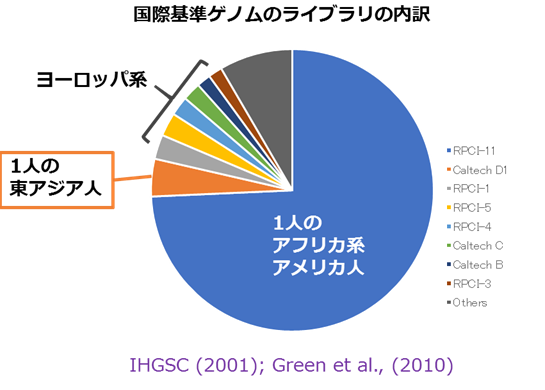
図1
今回公開したJG1の解析について
今回の解析においては、出身地域が異なる3名の日本人男性について、デノボアセンブリ*4、すなわち従来の国際基準ゲノムの配列情報を一切利用せずに全ゲノム配列の決定*5を行なった。デノボアセンブリには主に2種類のゲノム解読技術を用いた。一つは、ゲノムを1万塩基対程度の比較的長い単位(長鎖)で解読できる長鎖リード技術*6であり、もう一つは、解読したゲノム配列間を結合するための、オプティカルマッピング技術*7である。これらを3名それぞれについて行い、さらに短鎖技術による次世代シークエンシング解析を用いて解読エラーを除くことで、3名分の高精度な配列スキャフォールド*8を作成した。その上、メイトペアライブラリー技術*9を利用して3者間で解読できなかった領域を補いあい、3者の配列を統合した。その後3者で異なる部分は多数派の配列を採用することで、一個人に由来する配列バリエーションを取り除いた。最後に、既知の遺伝地図*10等の情報を用いて配列スキャフォールドとヒト染色体との対応付けを行い、日本人基準ゲノムJG1を完成させた(図2)。このようにして構築した日本人基準ゲノムJG1が上述のレアリファレンスアレル問題の解決に寄与するか検討するため、国際基準ゲノム配列とJG1を比較した。その結果、国際基準ゲノム配列には日本人集団には一切見られなかった配列バリエーション(一塩基多型)が246,464ヶ所で観察されたのに対し、JG1ではそのうちの241,500ヶ所を日本人全員に見られるアレルに置換していることがわかった。さらにJG1が日本人集団で多数派の配列バリエーションを多数有することは、日本人の次世代シークエンシング解析において短鎖リードの当てはめや配列バリエーション検出のエラーを劇的に減らし、より精密で正確なゲノム診断を可能にすることを示唆するものである。一塩基多型に限らずより大きな構造バリアントについても同様に多数派への置換を行なっているため、国際基準ゲノム配列を用いた解析に存在するバイアスを解決すると期待される。
なお、JG1は構成する3名のゲノムを複数の手法で高度に統合したものであるため、配列情報のみから3名の個人特定ができる可能性は極めて低い。

図2
今後の展望
今回構築に成功した日本人基準ゲノム配列JG1は、現在主流の一つである短鎖技術による次世代シークエンシング解析を、日本人を対象として行う場合に、国際基準ゲノム配列の代わりに利用することが可能になる。具体的には、以下のような利用が考えられる。
小児の希少疾患の原因解明
小児の希少疾患は、ライフステージの早期で発症することから環境要因よりも遺伝要因の効果が大きく、また、両親が同疾患に罹患していない場合には、両親からの継承ではなく、その配偶子形成時の新生突然変異が要因であると予想される。しかしながら、標準的なゲノム解析では、半数近くの症例家系で明確な原因突然変異は同定されていない。これは、一つにはゲノム解析の際に基準として用いた国際基準ゲノム配列に含まれるバイアスのため、そもそも原因となっている変異が解析できていない可能性がある。国際基準ゲノム配列に代えてJG1を用いることで、これまで見過ごされていた真の病因突然変異を同定することが期待される。我が国ではAMEDの進める「小児希少・未診断疾患イニシアチブ」(IRUD)などでの応用が考えられる。
日本人全ゲノムリファレンスパネルの再構築
ToMMoではこれまで、日本人全ゲノムリファレンスパネル3.5KJPNv2を最新として、日本人集団のゲノム配列バリエーションパネルを構築・公開してきた。これらは全て、国際基準ゲノム配列を基準とする短鎖技術による次世代シークエンシング解析によるものであった。JG1を基準ゲノム配列として再度、情報解析を行うことで、日本人全ゲノムリファレンスパネルに日本人集団のゲノム配列バリエーションをより高精度に収載できると期待される。
がんゲノムエクソーム解析への応用
がんゲノムのエクソーム解析(エクソンというゲノムの一部を解析する方法)によって、新規のがんドライバー変異や関連する配列バリエーションを発見しようという研究プロジェクトにおいても、中心的な技術は短鎖技術による次世代シークエンシング解析であることから、その基準ゲノム配列としてJG1を用いることで、高精度の解析が可能になると期待される。
ヒトゲノム突然変異率の推定
ヒトゲノム突然変異率とは、親から子へと世代間でゲノムが伝わる時にDNA塩基に突然変異が生じる割合のことであり、ゲノム医科学の基礎となる数値である。ヒトゲノム突然変異率の推定にあたっては、近年、短鎖技術による次世代シークエンシング解析を用いて親子間で全ゲノム配列を直接比較して推定する方法も試されているが、伝統的な手法である生物種の進化や分化の知見を用いて推定する方法との間で、未だ大きな食い違いがある。日本人の全ゲノム解析の基準ゲノムとしてJG1を用いることで解析の精度を向上させ、より正しい推定値を算出し、この矛盾を解決できることが期待される。ToMMoでは三世代コホート調査を行い親子のゲノムデータを収集していることから、JG1を基準とした解析により、本課題に取り組むことを検討している。
参考
- 東北メディカル・メガバンク計画について
- 東北メディカル・メガバンク計画は、東日本大震災からの復興と、個別化予防・医療の実現を目指しています。東北大学東北メディカル・メガバンク機構と岩手医科大学いわて東北メディカル・メガバンク機構を実施機関として、東日本大震災被災地の医療の創造的復興および被災者の健康増進に役立てるために、平成25年より合計15万人規模の地域住民コホート調査および三世代コホート調査等を実施して、試料・情報を収集したバイオバンク*11を整備しています。東北メディカル・メガバンク計画は、平成27年度より、国立研究開発法人日本医療研究開発機構(AMED)が本計画の研究支援担当機関の役割を果たしています。
- 未来型医療創成センターについて
- 未来型医療創成センター(INGEM)は、2018年3月、東北大学から東北大学病院、医学系研究科、加齢医学研究所、歯学研究科、薬学研究科、情報科学研究科、医工学研究科、そして東北メディカル・メガバンク機構の8つの部局が参画し、卓越した研究力を結集して未来型医療拠点の構築を目指し設立された組織です。
用語等説明・脚注
- ※1.国際基準ゲノム:
- 1990年代から2000年代にかけて行われた国際ヒトゲノム解読計画によって正確に塩基配列決定されたヒトゲノム配列。約30億塩基対からなる。現在の次世代シークエンシング解析のほとんどは、この配列を基準として参照のうえ行われている。
- ※2.ToMMoが実施した数千名の全ゲノム解析:
- 例えば、2018年6月にToMMoが構築を発表した日本人全ゲノムリファレンスパネル3.5KJPNv2は、3.5千人の全ゲノム解析をもとに、一塩基バリアント等の位置と頻度情報を公開している。国際基準ゲノム配列を参照配列として、短鎖技術による次世代シークエンシング解析を行った結果である。
- ※3.構造バリアント:
- ヒトゲノムを構成するDNA塩基一つ一つの変化ではなく、比較的多数の塩基がまとまりとなって、挿入・欠失したり、逆転したりしている部分。
- ※4.デノボアセンブリ:
- 国際基準ゲノム等の既知のゲノム配列に依らずに、シークエンサーにより解読したDNA断片の配列データから、元のゲノム配列を構築すること。
- ※5.国際基準ゲノムの配列情報を利用しない全ゲノム配列の決定:
- ToMMoでは、今回の成果に先立って、長鎖リード技術による解析で国際基準ゲノムに含まれていない日本人が持つゲノム配列情報を収集し、それを国際基準ゲノム配列に組み込んだ配列を、日本人基準ゲノム(JRG)として2016年以降公表している。
- ※6.長鎖リード技術:
- Pacific Biosciences社の一分子リアルタイムシークエンシング技術(SMRTシークエンシング技術)やOxford Nanopore社によるナノポアシークエンシング技術のこと。JG1の構築には、SMRTシークエンシング技術を用いて一検体当たり約3000億塩基対もの配列を解読した。このデータ量は、ヒトゲノム中の全ての領域を平均で100回ずつ解読したことになるため、理論的にはヒトゲノム全体を未決定な部分なくデノボアセンブリできる量となっている。
- ※7.オプティカルマッピング技術:
- 7文字程度の特定の塩基配列を蛍光ラベルしたDNA分子を微細な溝の中を通すことで直線状にし、蛍光ラベル間の距離を観測する手法。ゲノム中に複数回現れるこの塩基配列間の距離を記述した物理地図を作成しDNAシークエンサー技術で解読した配列を整列させることが可能である。本研究ではBionano Genomics社のオプティカルマッピング技術を利用した。
- ※8.配列スキャフォールド:
- 解読できた領域間を、未解読を意味する連続するNNN...Nで連結した塩基配列。ゲノム配列のうち解読できた断片はA,T,G,Cのいずれかの塩基シンボルで表現されコンティグ(シークエンサーで読み取られた配列を相同な重複部分で一つにつなげた配列)を形成する。物理地図や遺伝地図等の証拠で隣接することが判明したコンティグ同士の間をNNN...Nで連結し、スキャフォールドを作成する。
- ※9.メイトペアライブラリー技術:
- 短鎖技術では、通常千塩基以下のDNA断片の両端の数百塩基の配列情報が取得できる。メイトペアライブラリー技術を併用すると、数千から一万数千塩基のDNA断片の両端の塩基配列情報を、正確な向き情報とともに取得することが可能となる。このため、他の技術で解読した配列を整列することに利用される。
- ※10.遺伝地図:
- 染色体上の二点間の距離が大きいほどその間の組換えが起こりやすいことを利用して、観測された組換えの頻度から遺伝子やマーカーの並び順とその距離を記述したもの。連鎖地図とも呼ぶ。
- ※11.バイオバンク:
- 生体試料を収集・保管し、研究利用のために提供を行う。東北メディカル・メガバンク計画のバイオバンクは、コホート調査の参加者から血液・尿などの生体試料を集める。
お問い合わせ先
研究に関すること
東北大学東北メディカル・メガバンク機構
ゲノム遺伝統計学分野 教授 田宮元
電話番号:022-274-5996
生命情報システム科学分野 教授 木下賢吾
電話番号:022- 274-5952
報道に関すること
東北大学東北メディカル・メガバンク機構
長神 風二(ながみ ふうじ)
電話番号:022-717-7908
ファクス:022-717-7923
Eメール:f-nagami"AT"med.tohoku.ac.jp
AMED事業に関すること
国立研究開発法人日本医療研究開発機構
基盤研究事業部 バイオバンク課
電話番号:03-6870-2228
Eメール: tohoku-mm"AT"amed.go.jp
※Eメールは上記アドレス"AT"の部分を@に変えてください。
掲載日 平成31年2月25日
最終更新日 平成31年2月25日