


新たな先天奇形症候群の原因遺伝子を明らかに!―本疾患における転写制御因子のMN1遺伝子変異の同定―
プレスリリース
横浜市立大学
日本医療研究開発機構
横浜市立大学学術院医学群 遺伝学 三宅紀子准教授、松本直通教授らの研究グループは、中枢神経異常と特徴的な頭蓋顔面異常をきたす新たな先天性奇形症候群の原因となる遺伝子MN1を同定しました。この研究は同医学群 分子生物学 高橋秀尚教授、生化学 緒方一博教授、組織学 富澤信一講師、横浜市立大学先端医科学研究センター 木村弥生准教授、山形大学医学部附属病院 中村和幸医師、Centre Hospitalier Universitaire de Nantes Bertrand Isidor医師、広島市こども療育センター 平木洋子医師との共同研究による成果です。
研究成果のポイント
- 全エクソーム解析*1により、転写制御因子の一つであるMN1遺伝子の機能獲得型変異*2が、中枢神経異常と特徴的な頭蓋顔面異常をきたす新たな先天性奇形症候群を引き起こすことを明らかにした。
- MN1タンパク質は大部分が特定の構造を持たない天然変性領域*3から構成されており、相分離*4を呈する分子であることを明らかにした。
- MN1タンパク質の結合分子として転写因子であるPBX1, PKNOX1, ZBTB24と E3ユビキチンリガーゼの一つであるRING1を同定した。また、変異MN1タンパク質ではC末端が欠損しており、ZBTB24およびRING1との結合が阻害されていた。
- MN1タンパク質は、C末端にRING1が結合することでユビキチンプロテアソーム系*5による分解を受けるが、MN1変異体タンパク質はRING1との結合が阻害されることで分解を受けず、転写制御に異常をきたすことが本症候群発症のメカニズムの一因と考えられた。
研究の背景
研究の内容
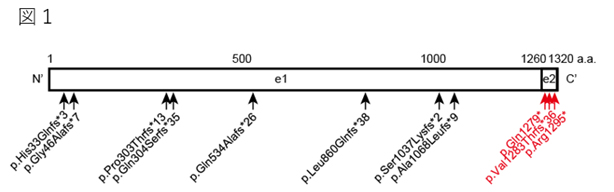
本研究グループは、精神・運動の発達遅延、言語障害、特徴的な頭蓋顔面異常(長頭症、平坦な顔、眼間解離、耳介低位、上向きの鼻孔など)、過食症、脳構造の異常を呈する新たな臨床像を呈する3症例を対象に、全エクソーム解析を用いて遺伝子変異の解析を行いました。その結果、3症例それぞれにMN1遺伝子のエクソン2に短縮型変異*8を認めました(図1)。
MN1遺伝子は二つのエクソン領域から構成されますが、健常者もしくはコントロール集団に認められた短縮型変異は、すべてエクソン1に存在しナンセンス変異依存mRNA分解 (NMD)*9を受けるため、MN1タンパク質が産生されないのに対し、患者に同定された変異はエクソン2にありNMDを受けないため、C末端の欠けた短いMN1タンパク質が産生されることが分かりました。もともとMN1タンパク質には細胞増殖を抑制することが知られていましたが、変異型を導入すると野生型よりも強い細胞増殖を抑制する効果が認められ、今回の変異が機能獲得型変異(細胞増殖抑制の増強)であることが分かりました。
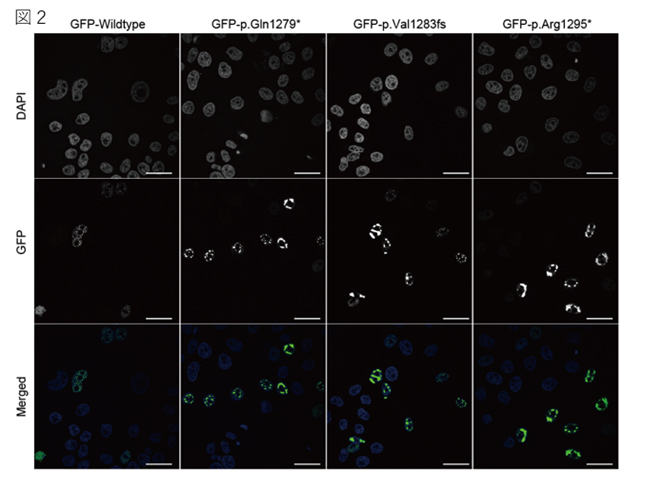
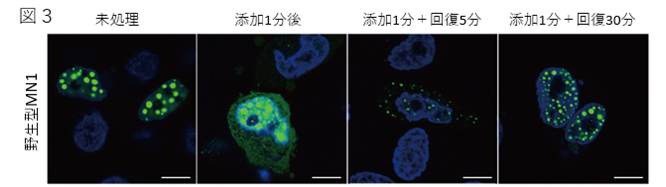
変異MN1タンパク質は、野生型と同様に転写活性を保持していました。しかし、野生型より安定して細胞内に存在し、核内で凝集体を形成しやすい傾向にありました(図2)。その凝集体が弱い疎水結合を阻害する溶媒(1,6-ヘキサンジオール)により可逆的に消失する(図3)ことから、MN1タンパク質が相分離を呈する分子であることを明らかにしました。MN1タンパク質は大部分が特定の三次構造を持たない天然変性領域から構成されており、C末端にある三次構造をとる領域の欠損により天然変性領域の占める割合が増えることと、タンパク質の安定性が増し細胞内での濃度の上昇により凝集体が増加することが考えられ、MN1タンパク質による転写制御に相分離が関与している可能性が示唆されました。
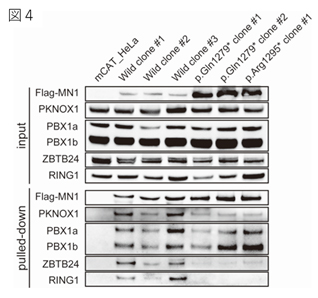
また、一連の機能解析により①MN1タンパク質が転写因子PBX1, PKNOX1,ZBTB24に結合すること(図4)、②MN1タンパク質は通常RING1を含むユビキチンプロテアソーム系により速やかに分解されていること、③ 変異MN1タンパク質では、ZBTB24とRING1との結合が阻害されていること(図4)が明らかとなりました。
以上より、本症候群発症のメカニズムとして、C末端が消失するMN1遺伝子の変異によりRING1との結合が阻害され、タンパク質の安定性が増し、通常のMN1タンパク質による転写制御のON, OFFのスイッチが適切に切り替わらないことで、下流の遺伝子の転写制御に異常をきたすことが考えられました。
今後の展開
今回、新たな先天性奇形症候群の原因となる遺伝子が同定されたことで、本症候群の病態解明と診断法・治療法の開発へ大きな寄与が期待されます。また、現在注目されているヒト疾患における相分離の役割の理解が進むことが期待されます。
用語説明
- *1 全エクソーム解析:
- ゲノム上のエクソン領域(遺伝子がタンパク質の配列を決定するゲノム中の領域)を網羅的に分画後、次世代シーケンサー(高速にゲノムの配列を読むことができる機械)を用いて塩基配列を決定する方法
- *2機能獲得型変異:
- 遺伝子産物が過剰な作用をする変異
- *3天然変性領域:
- 特定の構造をとらないタンパク質の領域のこと
- *4 相分離:
- 単一の均一混合物からの2つ以上の区別できる相が生成されること(液体ドレッシングが液滴化するように、もともとは同じ相を呈していたものが二つの相に分かれるような現象
- *5 ユビキチンプロテアソーム系:
- ユビキチンと呼ばれるたんぱく質に標識されたタンパク質は分解を受けるタンパク質の分解機構
- *6 転座:
- 異なる染色体の一部が切断、再結合して一部が入れ代わる変化
- *7 ノックアウトマウス:
- 人工的に遺伝子が機能しないようにしたマウス
- *8 短縮型変異:
- 合成されるたんぱく質が短くなるような変異
- *9 ナンセンス変異依存mRNA分解(NMD):
- 最終のスプライス部位の50-55塩基以上の上流に、中途型終始コドン(本来の位置よりも早い位置でタンパク質合成を終わらせるような終始コドン)が含まれるmRNA分子を特異的に分解する機構
※本研究は、米国の科学雑誌『American Journal of Human Genetics』に掲載されます。(米国東部時間12月12日午前11時付:日本時間12月13日午前1時付オンライン)
掲載論文
Gain-of-Function MN1 Truncation Variants Cause a Recognizable Syndrome with Craniofacial and Brain Abnormalities
Miyake N, Takahashi H, Nakamura K, Isidor B, Hiraki Y, Koshimizu E, Shiina M, Sasaki K, Suzuki H, Abe R, Kimura Y, Akiyama T, Tomiza S, Hirose T, Hamanaka K, Miyatake S, Mitsuhashi S, Mizuguchi T, Takata A, Oho K, Kato M, Ogata K, Matsumoto N.
The American Journal of Human Genetics(2020)
https://doi.org/10.1016/j.ajhg.2019.11.011
※本研究は、国立研究開発法人日本医療研究開発機構(AMED)の難治性疾患実用化研究事業「希少難病の高精度診断と病態解明のためのオミックス拠点の構築」(研究代表者:松本直通)、厚生労働省、文部科学省、科学技術振興機構、日本学術振興会、武田科学振興財団の研究補助金により行われました。
※本研究は、横浜市立大学大学院医学研究科 分子生物学 広瀬智威講師、佐々木和教助教、鈴木秀文助教、阿部竜太氏、遺伝学 輿水江里子研究員、濱中耕平研究員、三橋里美助教、水口剛講師、高田篤講師、組織学 大保和之教授、横浜市立大学先端医科学研究センター 秋山知子技術員、横浜市立大学附属病院 遺伝子診療部 宮武聡子講師、昭和大学 加藤光広教授、九州大学生体防御医学研究所 小田瑞穂氏、木庭絵美子氏、松本雅記准教授、中山敬一教授の協力を得て行われました。
お問い合わせ先
本資料の内容に関するお問い合わせ
公立大学法人横浜市立大学学術院医学群 遺伝学
教授 松本 直通
准教授 三宅 紀子
TEL:045-787-2606 FAX:045-786-5219
E-mail:naomat “AT” yokohama-cu.ac.jp
nmiyake “AT” yokohama-cu.ac.jp
取材対応窓口、詳細の資料請求など
公立大学法人横浜市立大学 研究企画・産学連携推進課長
渡邊 誠
TEL:045-787-2510 FAX:045-787-2509
E-mail:kenkyupr “AT” yokohama-cu.ac.jp
AMEDの事業について
国立研究開発法人日本医療研究開発機構 戦略推進部 難病研究課
TEL:03-6870-2223
E-mail:nambyo-info “AT” amed.go.jp
※E-mailは上記アドレス“AT”の部分を@に変えてください。
関連リンク
掲載日 令和元年12月13日
最終更新日 令和元年12月13日