


新技術ロングリード・シークエンサーで複雑なゲノムの構造変化を解明する手法を開発
プレスリリース
横浜市立大学
日本医療研究開発機構
横浜市立大学大学院医学研究科遺伝学三橋里美助教(現東京医科歯科大学難治疾患研究所准教授)、尾堀佐知子医師、松本直通教授らの研究グループは、ロングリード・シークエンサー*1を用いた新しいデータ解析手法を開発、これを応用し複数の症例で疾患に関わる複雑なゲノム構造異常を完全に解き明かしました。この研究で用いた新規解析手法は、国立研究開発法人産業技術総合研究所人工知能研究センターマーティン・フリス上級主任研究員、大阪大学微生物病研究所伝情報実験センターゲノム情報解析分野加藤和貴准教授との共同研究で開発されました。
本研究は、『Genome Medicine』に掲載されます。(日本時間 2020年7月31日午前9時付オンライン)
研究成果のポイント
- 新技術ロングリード・シークエンサーによる全ゲノム解析から、複雑なゲノム構造異常を迅速かつ効率的に検出する手法を開発
- 非常に複雑に絡み合ったゲノム構造の変化である染色体破砕*2(クロモスリプシス)の全体構造を明らかにした
- 新手法による複雑なゲノム構造変化の解明が、遺伝性疾患の病態解明に繋がることが期待される
研究の背景
遺伝性疾患の原因となるゲノム変化の解明には、DNA塩基配列*5を直接読む方法(DNAシークエンサー)や、マイクロアレイなどが汎用されてきました。遺伝性疾患の原因となるゲノム異常は、DNAの塩基変化(SNV:single nucleotide variant)と大きな領域のコピー数が変わる変化(CNV:copy number variant)が知られていますが、そのほかにも、染色体転座*3や、染色体の構造が複雑に変化する染色体破砕*2(クロモスリプシス、chromothripsis)と呼ばれる現象など、様々なものがあり、構造多型*6と呼ばれています。
従来、網羅的にDNA塩基配列を解析するにはショートリード・シークエンサーと呼ばれる200塩基程度の配列を一度に読むことのできる装置を使うことが一般的ですが、近年この100倍もの長さを読むことのできるロングリード・シークエンサーが実用化され、疾患のゲノム解析にも応用されるようになってきています。
長い配列を読むことができるようになると、染色体の広範な領域の構造変化をより正確に検出できるようになると期待されています。しかし、ロングリードのデータから、複数の個人での構造変化を比べることや、複雑な構造変化を検出することはまだ効率的手法が限られ容易ではなく、新しい手法の登場が待ち望まれていました。
研究の内容
従来の方法では解明が困難であった染色体転座を持つ先天奇形や卵巣機能不全等の症状のある患者さん4例について、ロングリード・ナノポアシークエンサーを用いて全ゲノム解析を行いました。本邦で開発された配列アライメント*7のデータ解析ツールであるLAST(LAST: genome-scale sequence comparison)を用いて、参照ヒトゲノム配列にアライメントを行い、共同研究により新規に開発したdnarrange(ディーエヌアレンジ)というソフトウェアを用いることで、構造変化を検出しました(図2)。
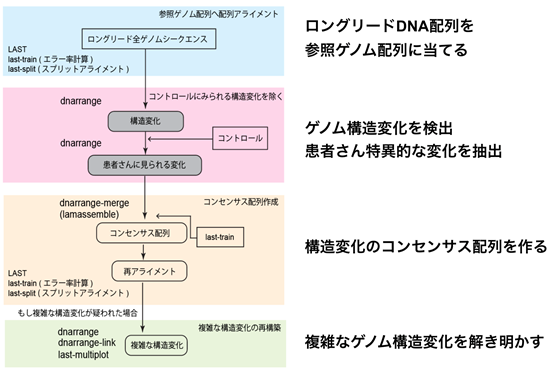
ヒトのゲノムには疾患を引き起こさない構造変化が数千個以上存在しています。これらをうまく除外することにより、疾患と関係する構造変化をより効率よく見つけることができると考えられます。今回、同じ疾患を持たない個人のゲノム(コントロール)に存在する構造多型を、dnarrangeを用いて除外し、患者さんだけで見られる病的意義を持つ候補となる構造異常領域を100個以下に絞ることができました。
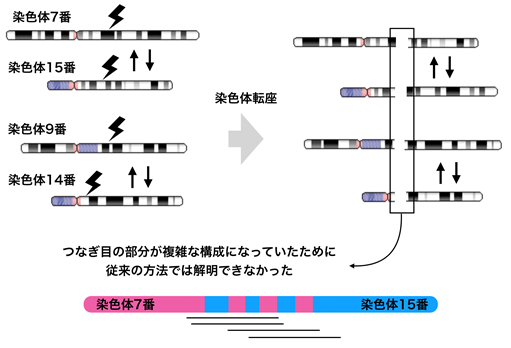
1例の患者さんでは、5本の染色体が18個の切断点を持ち、非常に複雑な構造変化をきたしていました。この患者さんのゲノムが実際にどのように変化しているのかを知ることは、従来法では極めて困難でした。そこで本研究では、これらの構造多型の位置関係を、自動で推測するツールdnarrange-linkを開発することにより、この複雑なゲノム構造異常を再構成することができました。この再構成によって、染色体破砕により3箇所の広い領域が欠失していることがわかりました(図3)。これらの欠失は、ゲノム構造を完全に再構成することで初めて描出することが可能となるため、従来の切断点を検出する手法だけで欠失したゲノム領域を同定することは困難であったと考えられます。
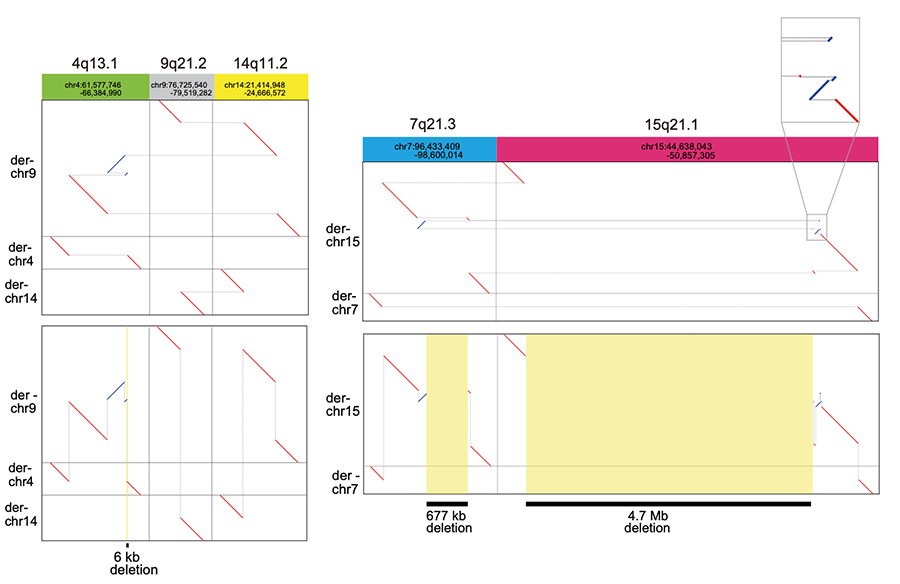
また、構造変化の影響やメカニズムを調べるには、ゲノム上の切断点を知ることが重要です。しかし、現在のロングリード・シークエンサーから出されるデータはエラーが多いため、一塩基の精度で正確な配列を知ることは困難な場合がありました。そこで多重アライメントソフトウェアMAFFT(MAFFT - a multiple sequence alignment program)を用いて、コンセンサス配列*9を作成する手法lamassemble(ラマセンブル)を開発し、構造変化部位のコンセンサス配列を得ました(図2)。コンセンサス配列から明らかになった構造多型の切断点は、サンガーシークエンス*10により確認された切断点と0~1塩基しかずれておらず、極めて高精度であり、染色体破砕をきたしたゲノム構造異常のメカニズムを考察することが可能となりました。
今後の展開
本研究により、従来の手法で解決困難であった染色体転座の患者さんのゲノム配列を、新技術であるロングリード・シークエンサーで読んだデータを用いて、疾患と関係するゲノム構造変化を明らかにする手法を確立しました。従来の手法では解析が難しい、染色体破砕のような非常に複雑な構造変化が解き明かされるようになり、遺伝性疾患の病態解明に繋がることが期待されます。
用語説明
- *1 ロングリード・シークエンサー:
- 1万塩基以上のDNA配列を一つづきに読むことができる装置。Oxford Nanopore TechnologiesやPacific Biosciences社の装置が使われる。
- *2 染色体破砕(クロモスリプシス):
- 染色体が複数の断片に破砕され、再結合されることでゲノムに複雑な構造変化が起きること。遺伝子領域が欠損したり、遺伝子の発現に影響を与えたりすることで、がんや先天性疾患を引き起こすことが知られている。
- *3 染色体転座:
- 染色体の一部が切断され、異なる染色体と再結合して入れ代わる変化。
- *4 ナノポアシークエンサー:
- Oxford Nanopore Technologies社のロングリード・シークエンサー。ナノポアというタンパク質の穴が人工膜に埋め込まれており、ナノポアをDNA分子が通り抜ける時に起きる電流の変化によって、DNAの配列を解析することができる。
- *5 DNA塩基配列<:
- ゲノムを構成するDNA塩基配列は、グアニン(G)、シトシン(C)、アデニン(A)、チミン(T)の4種類からなる。
- *6 構造多型:
- ゲノムの比較的大きな領域に起きる変化。欠失・重複・逆位・転座・挿入など、複数のパターンに分類される。量の変化を伴うものは、CNV(copy number variation)と呼ばれる。
- *7 配列アライメント:
- 複数のDNAなどの配列を、似たものが同じ位置に来るように並べたもの、またその手法。
- *8 解析パイプライン:
- データ解析は複数の手法を使って行われることが多いが、これらを一連の流れとして組み合わせたもの。
- *9 コンセンサス配列:
- 配列アライメントの各位置において最も確からしい塩基を計算し、1種類の配列を決めること。
- *10 サンガーシークエンス:
- 1970年代にフレデリック・サンガーが開発した塩基配列の決定法で、DNAポリメラーゼを用いて、末端が特定の塩基に対応するDNA断片を合成し、一つひとつDNA配列を決めていく。
掲載論文
A pipeline for complete characterization of complex germline rearrangements from long DNA reads
Satomi Mitsuhashi, Sachiko Ohori, Kazutaka Katoh, Martin C Frith, Naomichi Matsumoto
Genome Medicine (2020)
- DOI:
- 10.1186/s13073-020-00762-1
研究支援
※本研究は、国立研究開発法人日本医療研究開発機構(AMED)の難治性疾患実用化研究事業「新技術を用いた難治性疾患の高精度診断法の開発」(研究代表者:松本直通)、厚生労働省、日本学術振興会、武田科学振興財団の支援により実施されました。
※本研究の共同研究により開発されたツールは以下になります。
dnarrange:https://github.com/mcfrith/dnarrange
lamassemble:https://gitlab.com/mcfrith/lamassemble
お問い合わせ先
本資料の内容に関するお問い合わせ
公立大学法人横浜市立大学大学院医学研究科 遺伝学 教授 松本直通
TEL:045-787-2606 FAX:045-786-5219
E-mail:naomat“AT”yokohama-cu.ac.jp
取材対応窓口、詳細の資料請求など
公立大学法人横浜市立大学研究・産学連携推進課長 山﨑理絵
TEL:045-787-2510
E-mail:kenkyupr“AT”yokohama-cu.ac.jp
AMEDの事業について
国立研究開発法人日本医療研究開発機構
ゲノム・データ基盤事業部医療技術研究開発課
TEL:03-6870-2221
E-mail:nambyo-r“AT”amed.go.jp
※E-mailは上記アドレス“AT”の部分を@に変えてください。
関連リンク
掲載日 令和2年7月31日
最終更新日 令和2年7月31日