


細胞の老化が阻害されてがんが発生する仕組みをハエで解明―マイクロRNAが細胞老化遺伝子を破壊してがん化を促進―
プレスリリース
京都大学
日本医療研究開発機構
概要
がんは「がん遺伝子」の活性が高まることで引き起こされますが、それだけではがんは生じません。なぜなら、がん遺伝子が活性化すると細胞の増殖が促されるとともに「細胞老化1」と呼ばれる現象が起こり、細胞の増殖を止めようとする働きが生まれるからです。つまり、細胞老化はがんの発生を防ぐバリアとして働いており、この機能がなくなるとがんの発生が促されます。しかし、がんが発生する際にどのようなメカニズムで細胞老化の機能が抑制されるのかはよくわかっていませんでした。
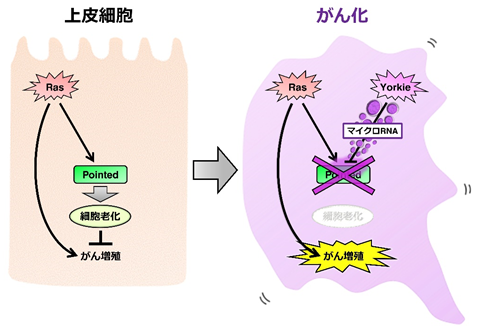
今回、京都大学大学院生命科学研究科 井垣達吏 教授、井藤喬夫 研究員の研究グループは、ショウジョウバエを用いてがんの発生メカニズムを解析する中で、ある特定の「マイクロRNA2」が細胞老化を阻害し、がん化を促すことを発見しました。ヒトの多くのがんで活性化しているがん遺伝子「Ras3」は、細胞老化を引き起こすことが知られています。ショウジョウバエの複眼でRasを活性化してもがん化は起こりませんが、がん促進タンパク質「Yorkie4」(ヒトではYAP)を同時に活性化させると激しいがん化が起こることがわかりました。そのメカニズムとして、YorkieがマイクロRNAの発現を導き、これが細胞老化を引き起こすために必要な「Pointed5」(ヒトではETS)と呼ばれる遺伝子のメッセンジャーRNAを破壊することで細胞老化が起こらなくなり、がん化が促進されることがわかりました。今回明らかになった細胞老化の制御メカニズムを標的として、新たながん治療法を開発できる可能性があります。
本研究成果は、2021年6月1日に米科学誌「Science Signaling」のオンライン版に掲載されました。
背景
細胞老化とは、細胞の細胞周期が不可逆的に停止して、細胞増殖が起こらなくなる現象です。細胞老化は様々な細胞内ストレスによって誘導されますが、その1つが「がん遺伝子の活性化」です。一見矛盾しているようですが、がん遺伝子の活性化は細胞の増殖を促す一方で、細胞老化を引き起こしてがんの発生を防いでいます。これは、多細胞生物が進化の過程で細胞増殖の仕組みを複雑化するのにともなって、個体のがん化を防ぐために獲得した防御メカニズムである可能性が考えられます。多くのがんの発生にはがん遺伝子の活性化が関わっていることから、がんが発生するにはその過程で細胞老化が阻害されると考えられます。しかし、がん遺伝子を活性化した細胞がどのようにして細胞老化を免れてがん化していくのか、そのメカニズムはあまりわかっていませんでした。
マイクロRNAとは、タンパク質を作りださない短鎖(20-25塩基)のRNA(ノンコーディングRNA)です。マイクロRNAは、自身と同じ配列をもつ遺伝子のメッセンジャーRNA(mRNA)に結合し、そのmRNAを分解したりタンパク質への翻訳を阻害したりすることでその遺伝子の発現を抑制します。ヒトの様々ながんでマイクロRNAの発現量が異常になっていることが報告されていることから、がんの発生にマイクロRNAが深く関わっている可能性が考えられています。しかし、マイクロRNAがどのようにがんの制御に関わっているのか、そのメカニズムについてはあまりわかっていません。
今回、ある特定のマイクロRNAが細胞老化を阻害することでがん化を促進することを見いだし、そのメカニズムの詳細を明らかにすることに成功しました。
研究手法・成果
本研究では、ショウジョウバエをモデル生物として用い、細胞のがん化を促進する遺伝子変異を探索しました。具体的には、まずハエの複眼の上皮組織でがん遺伝子Rasを活性化させ、小さな腫瘍を誘導します。この時、同時に様々な遺伝子を一つ一つ欠損させ、がん化が強く促進されるものを探索しました。その結果、Pointedと呼ばれる転写活性化因子(ヒトではETSと呼ばれる)を作りだす遺伝子の発現を抑制すると、がん化が著しく亢進することがわかりました。さらに解析を進めた結果、PointedはRasの活性化によりその発現が誘導され、細胞老化を引き起こす働きをもつことがわかりました。つまり、Rasを活性化した細胞内でPointedの発現が抑制されると細胞老化が起こらなくなり、細胞のがん化が強く促進することがわかりました。
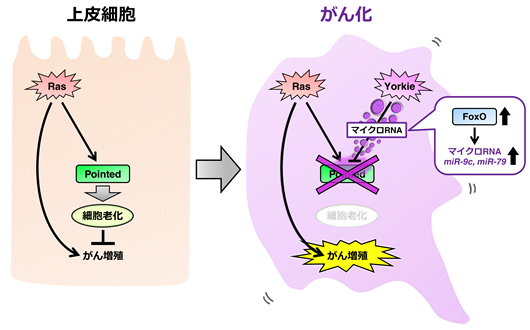
次に、ショウジョウバエの発がんモデルを用いて、発がん過程でのPointedの働きを解析しました。がん化の原因の1つとして、上皮細胞の頂端-基底極性(細胞極性)の崩壊が知られています。ショウジョウバエの複眼でRasを活性化し、同時に細胞極性を破壊すると、激しく増殖して浸潤・転移する悪性腫瘍(がん)が形成されることが知られています。興味深いことに、この悪性腫瘍ではPointedの発現が抑制され、細胞老化が阻害されていることがわかりました。そのメカニズムを解析した結果、極性を失ったRas活性化細胞では転写共役因子Yorkie(ヒトではYAPと呼ばれる)が活性化しており、Yorkieの活性化がFoxOと呼ばれる転写因子の発現量を増加させることがわかりました。このFoxOがmiR-9cおよびmiR-79と呼ばれる2種類のマイクロRNAの発現を誘導し、これらのマイクロRNAがPointedのmRNAを破壊することでPointedの発現が抑制され、細胞老化が起こらなくなることがわかりました(参考図)。
波及効果、今後の予定
今回明らかになった細胞老化の阻害メカニズムに関わる因子群は哺乳類においても保存されているため、ヒトのがんでも同様のメカニズムが機能している可能性が考えられます。また、ヒトの様々ながんでRasの活性化、細胞極性の崩壊、YAPの活性化、そしてmiR-9(miR-9cに相当するマイクロRNA)の高発現が見られることが報告されています。これらのことから、今回の研究成果が新たながん治療法の開発に貢献することが期待されます。また、細胞老化は個体の老化や寿命にも密接に関わっている現象のため、今回見いだしたメカニズムが抗老化に関わる医学分野へ応用されることも期待されます。
研究プロジェクトについて
本研究は、AMED「老化メカニズムの解明・制御プロジェクト」の支援のもとで行われました。
論文タイトルと著者
- タイトル
- Yorkie drives Ras-induced tumor progression by microRNA-mediated inhibition of cellular senescence(Yorkieの活性化はmicroRNAを介した細胞老化の抑制によりRas活性化腫瘍の悪性化を引き起こす)
- 著者
- Takao Ito and Tatsushi Igaki
- 掲載誌
- Science Signaling
- DOI
- 10.1126/scisignal.aaz3578
用語解説
- 1.細胞老化
- 細胞が不可逆的に細胞分裂を停止する現象。ショウジョウバエから哺乳類まで進化的に保存されている。様々な細胞内ストレスによって引き起こされる。
- 2.マイクロRNA
- 短鎖のノンコーディングRNA。標的配列を介して標的遺伝子のmRNAを分解する。
- 3.Ras
- ヒトのがんの約30%において活性化しているがん遺伝子。すい臓がんではその約90%で活性化している。
- 4.Yorkie
- 転写共役因子。細胞増殖の促進と細胞死の抑制によって細胞の異常増殖をもたらす。
- 5.Pointed
- 進化的に保存されたETSファミリーに属する転写活性化因子。PointedのヒトホモログであるETS1とETS2は、CDK阻害因子p16の発現誘導を介して細胞老化制御に関与することが報告されている。
お問い合わせ先
井垣達吏(いがきたつし)
京都大学大学院生命科学研究科・教授
Tel:075-753-7684(教授室)
Fax:075-753-7686
E-mail:igaki.tatsushi.4s“AT”kyoto-u.ac.jp
報道・取材に関するお問い合わせ
京都大学 総務部広報課国際広報室
Tel:075-753-5729
Fax:075-753-2094
E-mail:comms“AT”mail2.adm.kyoto-u.ac.jp
AMEDに関するお問い合わせ
日本医療研究開発機構(AMED)
疾患基礎研究事業部 疾患基礎研究課
Tel:03-6870-2286
Fax:03-6870-2243
老化メカニズムの解明・制御プロジェクト
E-mail:aging“AT”amed.go.jp
※E-mailは上記アドレス“AT”の部分を@に変えてください。
関連リンク
掲載日 令和3年6月9日
最終更新日 令和3年6月9日