


アーカイブ 日本人の多因子疾患関連遺伝子の同定を目指し、大規模全ゲノム情報作成のための解析手法を開発する

河合 洋介
東京大学大学院医学系研究科 特任助教
ヒトゲノムプロジェクトが2003年に完了して以降、個人の一塩基多型(Single Nucleotide Polymorphism:SNPs)をSNPアレイなどの新たなテクノロジーによってゲノム規模で調べられるようになった。そして、比較的身近な疾患に共通するSNPsなどの遺伝子変異を全ゲノム領域にわたって関連解析するゲノムワイド関連解析(Genome-Wide Association Study:GWAS)が盛んになり、すでに、さまざまな疾患の感受性遺伝子が同定されている。一方で、次世代シーケンサー(Next Generation Sequencer:NGS)による全ゲノム解析も飛躍的に進んでいる。 「多くのサンプルの解析に長けたSNPアレイと、あらゆるタイプの多型情報を高精度に解析できるNGS、この両者の長所を活かした解析手法を開発し、これまでに蓄積したデータだけでなく、今後得られるデータにも使えるようにするのが目標です」と河合 洋介先生は話す。
SNPアレイとNGS、2つの性質の異なるゲノム情報を統合し、これまでGWASではわからなかった遺伝子の多型や変異を明らかにできれば、さらに多因子疾患の遺伝的要因の解明が進むと期待されている。
河合先生は、2017年8月に東京大学大学院医学系研究科人類遺伝学教室に着任し、平成28年度ゲノム医療実現推進プラットフォーム事業「先端ゲノム研究開発」(GRIFIN)プロジェクト「日本人大規模全ゲノム情報を基盤とした多因子疾患関連遺伝子の同定を加速する情報解析技術の開発と応用」(2016年採択)のメンバーとして挑戦中だ。このプロジェクトでは、すでにある日本人の健常者の全ゲノムリファレンスパネルや多因子疾患パネルを活用かつ拡大しながら、疾患の遺伝的要因をゲノム情報から探索するための情報解析手法を開発し、応用している。
SNPアレイとNGSのデータを比較し、解析して、見えていないSNPの情報を推定する
GWASでは60万ほどの塩基対の解析が行われる一方、NGSならば2,000~3,000万のSNPsを一度の解析で調べられる。データベース上にはこれまでに蓄積されたさまざまな個人や集団のデータが混在していて、ある特定の特定個人のSNPsとデータベース上のデータにゲノム全体がぴたりと重なることはほとんどあり得ないが、河合先生によると「人類のゲノムは遺伝的な多様性が意外と低く、同じ集団の解析で得られたSNPデータの中には個人間で共通した塩基の並び(ハプロタイプ)を見つけ出すことができる」という。そこで、NGSのデータとGWASで得られたSNPアレイのデータを比較して、SNPアレイには存在しないSNPsの遺伝型をNGSのデータから推定することができる。これがジェノタイプインピュテーションの原理である(図1)。この手法を応用することによって、これまでに蓄積した大量のGWASデータからNGS解析に匹敵する多型情報を得られ、過去の研究で見逃していた疾患発症の遺伝的な要因が発見できるようになるというわけだ。
このプロジェクトでは、SNPのインピュテーションだけではなく、SNPアレイでは調べることができない遺伝子のコピー数多型(Copy Number Variation:CNV)や反復配列(Short Tandem Repeat:STR)のインピュテーションやその結果を利用したGWAS手法、日本人の遺伝的な多様性を考慮した解析手法を開発し、基盤情報を作成することによって、新規疾患関連変異をより多く、かつ、精密に同定することを目指している。
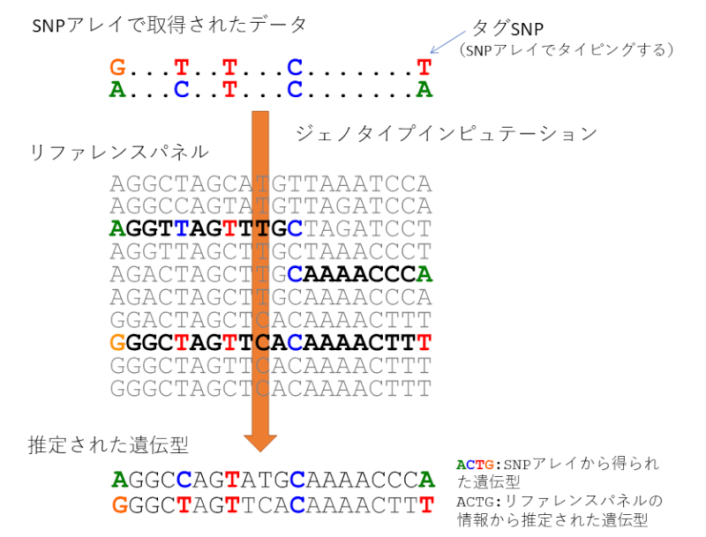
図1:SNPアレイによるジェノタイプインピュテーションの概念図
河合先生によると、特にHLA遺伝子の研究は近年大きく進展しているという。HLA型は、ヒトの体が自己を認識するのに使う部分であることから、この進展の成果は、自己免疫疾患においては大きな意味を持つ。「疾患一般では患者と健常者のオッズ比は高くて2倍ほどですが、自己免疫疾患では20倍くらいまで跳ね上がる場合があります。個人のHLA型を血液型のように調べて、自己免疫疾患などの発症予測や薬の副作用の予測、治療の効果アップの判定に使う時代が来るのではないでしょうか」と河合先生は予想している。一方、HLAは遺伝子座内に多数の遺伝子が存在するため、これまでの解析には各HLA遺伝子を血清で調べるという実験的なアプローチが必要だった。それがNGSやSNPアレイのデータに特定のアルゴリズムを与えるHLA型推定(HLAインピュテーション)によって、複数のHLA遺伝子の型を一度に決められるようになり、過去のデータに対しても実験をせずにHLA型推定を行えるようになったのだ(図2)。(関連サイト情報1参照)
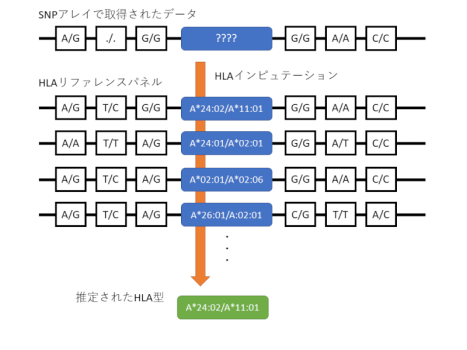
図2:インピュテーションの概念図
河合先生らが行った研究例として、薬の激烈な副作用として知られるスティーブンス・ジョンソン症候群(以下、SJS)がある。この疾患は、唇や口腔内、目や外陰部などの粘膜がただれ、高熱や全身倦怠感が出て、重症化すると死に至る重大なもので、国の難病指定を受けている。
「この研究では、東北メディカル・メガバンク機構(ToMMo)が開発した、日本人ゲノム用SNP解析ツール『ジャポニカアレイ』を用いてインピュテーションを行うことによって、これまでに関連が知られていたHLA遺伝子だけではなく、新たなSJSに関連するSNPを発見することができました」(関連サイト情報2、関連サイト情報3参照)
肝臓の中の極細い胆管が壊れ、胆汁の流れが滞り、肝臓の働きが低下する原発性胆汁性胆管炎でも、新たな成果が出た(図3)。国の指定する難病であるこの病気はHLA型が関連する自己免疫疾患であることがわかってきたが、HLA型の変化だけでは説明できない。そこで、河合先生の参加する研究チームは新たな感受性遺伝子を同定するためにジェノタイプインピュテーションを行い、3つの遺伝子領域から新たなSNPを発見し報告した。 (関連サイト情報4参照)現在は、ナルコレプシーやB型肝炎、パニック障害、炎症性腸疾患などでの研究を続けている。
研究では、このプロジェクトの研究代表者で同教室の徳永勝士教授らが国内外でこれまでデータを積み上げてきたヒトゲノムバリエーションデータベース (「SNP-based GWASデータベース」、「標準CNVデータベースおよびCNV based GWAS データベース」、「HLA データベース」、「Human Variation データベース」)に格納されたデータや未発表のGWASデータ、およびToMMoなどの持つ全ゲノムデータベースを主に活用している
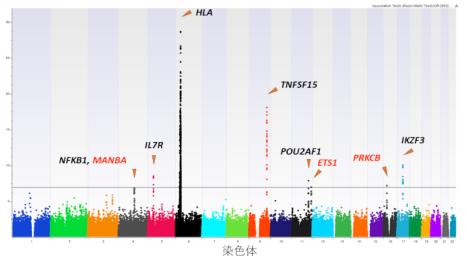
図3:原発性胆汁性胆管炎(PBC)のGWASの結果SNPアレイデータにジェノタイプインピュテーションを行って解析したもの。赤字部分が新たに見つかった関連遺伝子。
さまざまな専門性を持つチームで、異質なデータを解析する技術を開発する
このプロジェクトを進める上で苦労する点は、検体を提供した健常者や患者のグループ・地域、SNPアレイやNGSの種類、解析方法、出力フォーマットなどが異なるデータを扱うため、統合にあたって組み合わせを考え、使いやすい形に変える必要があること、また、データサイズが大きく、計算に使う東北大学のスーパーコンピューターの容量を圧迫することだという。「研究を効率化する方法を、常に工夫しなければならなりませんね」

ことに、研究の手応えを
感じているという。
GWASが抱える「失われた遺伝率」という根本的な問題もある。「多因子疾患は数十から、多ければ数千の遺伝子が関わると考えられていて、GWASの1,000~2,000人といった母数では、すべての遺伝子を見つけられません。もちろん世界中の研究者とのコンソーシアムでデータを集めれば精度は上がりますが、それでも100%ではありません」。河合先生は、この問題を氷山に例える。氷山の海面上に出ているところ、すなわちGWASで明らかになっているSNPsのデータから遺伝率は計算できても、実際は海面下にどういったデータが、どれくらい隠れているのはわからない。これが「失われた遺伝率」だ。「インピュテーションの方法の進展で、見えなかったSNPsやコピー数のバリアントが推定できるようになり、失われた遺伝率の解消ができるようになってきました。ただ、どこまでいけば終わりかはわからないのです」と河合先生。今後も新しい手法の開発も含めて、地道に不明な部分を推定し、検証していく作業が必要になる。
それでも「我々のプロジェクトチームの強みはメンバーに人類遺伝学、機械学習、実験などいろいろなバックグラウンドの専門家が揃っていて、お互いを補完し合っていいものを作っていけるところです。既存のインピュテーションの手法を検証するだけでなく、新たなインピュテーションの手法も開発したい。若手研究者や博士課程の学生も育てていきます」と意欲を燃やす。
もともと進化学に興味があり、植物や霊長類の起源に関する遺伝学的なアルゴリズムを作っていた河合先生。今の研究は「遺伝学の基礎や情報科学の理論が、臨床での治療や予防といった応用につながるまでの距離が短いところが魅力の1つ」と話す。「直接患者を救うことはなくても、役に立つ仕事になると思うので、やりがいがあります。GWASが示す免疫応答性や薬剤応答性のデータに人工知能や機械学習を組み合わせたハイブリッド型の日本人の全ゲノム情報を明らかにし、多因子疾患の発症リスクや創薬の標的を定める基盤を作ることで、ゲノム医学の研究推進や個別化医療の実現につなげたい」と笑顔で抱負を語った。
インタビュー動画
- コラム研究者紹介(youtube動画)
推薦論文
An integrated map of genetic variation from 1,092 human genomes
研究者経歴
1978年、岡山県生まれ。2001年に東京理科大学 理工学部 応用生物科学科卒業。2006年に同大学院理工学研究科応用生物科学専攻修了(博士(理学))。国立遺伝学研究所・特任研究員、立命館大学・助教、University of Washington・客員研究員、前橋工科大学・研究員、東北大学・講師を経て、2017年8月より現職。 専門は集団遺伝学・分子進化学。
関連リンク
掲載日 平成29年12月18日
最終更新日 令和2年3月30日