


アーカイブ 臨床ゲノムデータベースで切り拓く、難聴の原因遺伝子診断と個別化治療への道

西尾 信哉
信州大学医学部 人工聴覚器学講座 特任講師
先天性難聴は、新出生児1,000人に1人に認められる最も発症頻度の高い先天性障害の1つだ。(参考資料1)「先天性難聴の発症要因として一番頻度の高いのは遺伝子が関与するケース。おおよそ6~7割ぐらいであることが報告されています。難聴の原因遺伝子としては、現在までに約100種類の原因遺伝子が同定されており、非常にヘテロジェナイエティ(遺伝的異質性)の高い疾患です。異なった原因遺伝子が同じ難聴という症状を呈するため、臨床症状だけから原因遺伝子を推定することは非常に困難です。」と西尾 信哉先生は語る。
臨床所見や進行状況だけでは原因を"特定"することが難しいため、原因を明らかにするためには遺伝学的検査(遺伝子診断)が有効だ。見出された変異を照らし合わせるためのデータベースはデータ件数が多いほど診断に貢献する。検査コストを抑えながら、診断精度の高い検査方法を開発し、保険収載として臨床に還元すること。そして、米国・ゲノム変異情報データベース「ClinVar(クリンバー)」日本版を目ざした先天性難聴の臨床ゲノム大規模データベースを構築すること。その両輪は、難聴における診断から治療への架け橋づくりに欠かせない。
ターゲットリシークエンス解析の臨床応用が鍵 ~診断精度を高めるために


西尾先生は、宇佐美 真一先生が教授を務める信州大学医学部耳鼻咽喉科学教室(以下、宇佐美研)で次世代シークエンサー(Next Generation Sequencer:NGS)による難聴患者の遺伝子解析に取り組み、宇佐美先生らとGJB2遺伝子、SLC26A4遺伝子、CDH23遺伝子、OTOF遺伝子など非常に多くの原因遺伝子変異を特定する(図1:参考資料2)とともに、原因遺伝子の種類によって症状が異なることを明らかにしてきた(参考資料3、4)。また、解析したクリニカルシークエンス・データと臨床情報とを統合管理するデータストレージを開発してきたが、これをさらに充実、拡大しながら、診断のついたものをAMEDが用意する「臨床ゲノム統合データベース」(図2)に追加していくことが、平成28~30年度 臨床ゲノム情報統合データベース整備事業「感覚器障害領域を対象とした統合型臨床ゲノム情報データストレージの構築に関する研究」(研究開発代表者:信州大学 宇佐美 真一)での使命だ。データストレージに集積されるゲノム情報と臨床情報が増加するにつれ、原因遺伝子変異の候補を効率的に"絞り込む"ことが可能になったという。また、絞り込んだ候補の中から難聴の原因を同定できるケースも増加している。2018年8月現在、ゲノム解析が終了し登録された件数は、約8,000家系分にも及ぶ。
宇佐美研では、多数の原因遺伝子が関与する遺伝性難聴の診断を効率的に進めるために、日本人難聴患者の原因として頻度の高い13遺伝子46変異をインベーダー法で網羅的にスクリーニングする手法を開発し臨床応用してきた。この検査手法は、2012(平成24)年に「遺伝学的検査(先天性難聴)」として保険収載され、全国の大学病院などで日常の臨床検査ツールとして定着している。(この成功要因等についてはAMEDが調査を行いレポートとして「ゲノム医療実用化導入還元モデルに関する調査」(参考資料5)でまとめている)インベーダー法自体は、既知遺伝子変異の有無を調べる検査法として「国際 HapMap プロジェクト」においても採用された検査手法であり、定温でできるため簡便かつ効率的で、正確性も高い。
「もともとは、サンガーシークエンスで遺伝子を1個1個の調べることしかできなかったのですが、調べてみると原因遺伝子変異のうち、民族特異的に多い原因遺伝子変異があることわかってきたのです」(参考資料6)。そこで、日本人に多く見つかる原因遺伝子変異にターゲットを絞ることで検査にかかる所要時間とコストを削減し、検出率向上にも大きく貢献した。検査方法を開発した当時は、速く、コスト面でも優れていたインベーダー法だが、1SNP追加するごとに解析にかかる費用が増すという欠点があった。
インベーダー法を用いても原因遺伝子変異を特定できない場合、検査対象に含まれていない原因遺伝子や遺伝子変異が関与している可能性がある。これをどのように突き止めるか?
拡張性に限界が見え始めたことから、共同研究先企業とともに、難聴の原因遺伝子を網羅的に解析可能なNGS「Ion PGM」での検査を併用した難聴遺伝子診断の研究開発を進め、2015年8月より保険診療の検査でも同手法が用いられている。「検査として臨床応用するためにはデータの均質性、精度が重要。宇佐美研では対象となる遺伝子の96%以上の領域が20回以上読めていることを品質基準にしています。NGSで得たデータは、インベーダー法の結果と比較して99.98%一致しており、十分に臨床応用できる精度が得られました」と西尾先生は新たな診断方法の有用性に注目する。実際、保険の検査では、この新しい遺伝子解析手法を用いて解析対象を19遺伝子154変異まで増加させることで、変異検出率が31%から41%まで向上した(参考資料3)。「保険の検査では19遺伝子154変異の結果をお返ししていますが、解析自体は過去に難聴の原因として報告のある63遺伝子を全て調べています。今後、臨床的意義の明らかになった変異から順次保険の検査にフィードバックしていくことで、さらなる診断率の向上が期待できます」と語る。
また、保険の検査で利用しているNGSの機種に合わせてコピー数変化を解析する手法を独自開発し、診断率を2~3%向上させた。西尾先生は「全遺伝子解析の場合にはコピー数変化を測るのは比較的簡単です。しかし、100個の遺伝子だけを抜き取ったところからコピー数を調べるいい方法がなくて。私たちはマルチプレックスPCRでターゲット領域を増やしたものをNGSで読むのですが、そもそもPCRでは増幅しやすいプライマーとそうでないものがありますので、増幅効率にバラつきが出ます。自主製作したプログラムでは、そこをうまく補正しているのです」(参考資料7)。
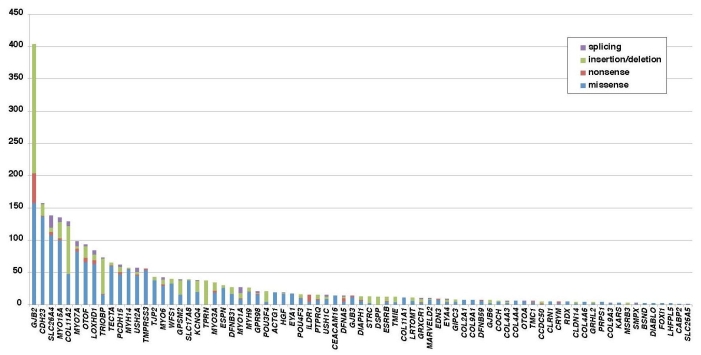
図1:日本人難聴患者1,120人から見出された難聴原因遺伝子変異。(参考資料2)
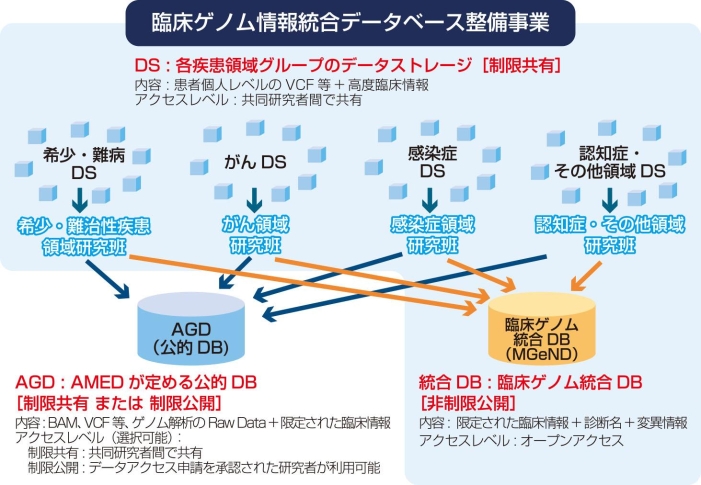
図2:臨床ゲノム情報統合データベース整備事業 概要図
数は力に。診断から治療への展望と課題
先天性難聴は基本的にメンデル遺伝性疾患であるため、変異を持っていれば必ず発症するし、持っていなければ発症しない(常染色体劣性遺伝形式の場合には両アリルに変異がある場合に発症する)。しかし、1人の患者を対象に、過去に難聴の原因として報告のある100遺伝子を全て解析すると1,000個にも及ぶ遺伝子変異が見つかる。全て過去に難聴の原因として報告のある遺伝子から見つかった変異であるため、その中から難聴の原因となっている1個を見つけ出すには豊富な経験と症例の蓄積がなければ難しい。それを誰が見てもわかる状態にしたのが、宇佐美研のデータストレージの凄さだ。家系図、難聴のタイプや重症度、その他の臨床情報、本人と両親より見出された遺伝子変異が分かりやすく表示される(図3)。
もう1つの強みは、変異のナレッジベースも用意されていることだ(図4)。「この変異が、どのぐらいタンパク質の機能や立体構造に影響を及ぼすかを予測するスコア、それがグラフでぱっと表示されます。また、過去に報告のある変異かどうかも一目でわかるようになっています。さらに、1,000人ゲノムなどの公的コントロールデータベースに加え、東北大学東北メディカル・メガバンク機構(以下、ToMMo)の3,500人分のゲノムと京都大学の1,200人分のエクソームの中でどの程度の頻度で見つかるかを瞬時に調べることができます。日本人のコントロールデータがあるということは、特にパワフルで、今後ますます期待できるところです。数は力(ちから)です。ToMMoとは共同研究契約を結び、一般には公開していない欠失・挿入変異のデータを分譲してもらって活用しています。あと、私たちがもつ難聴患者約8,000家系の中で同じ変異を持っている患者が何例いるか、それはコントロールと比べて有意差があるかどうかもわかります。有意差がある場合には、さらにオッズ比がどのぐらいあるかも。これらを自動計算するようにプログラムを組んであります。さらに同じ変異を持つ患者同士を相互に比較し、臨床的特徴が類似しているかも簡単に比べることができます」と西尾先生はモニター上に素早くデータを表示させながら説明した。保有するクリニカルシークエンス・データ数の多さと国内バンクとの連携により、非常に効率的に診断を行うためのシステムが完成したといえるだろう。

難聴データストレージを操作する様子。
このデータストレージのシステムはFileMaker Proで構築されている。データベースエンジンを選定する際、業界標準のPHP+MySQLも検討したが、臨床医が容易に操作できること(特に各種検索機能の容易さ)、インターフェース作成・改変の柔軟性の高さ、容易に各種プラットフォーム(Mac、Win、タブレット端末)に対応可能な点などを考慮し、決めたという。また、このシステムが他の疾患にも応用できると考え、2017年度に論文として発表するとともにアカデミアには無償提供を行なっている(参考資料8)。
データストレージを運用し、年間約1,200家系分の解析データと臨床情報を集積するとともに、新規の変異が見つかれば家系解析やモデル動物で病原性確認し保険にフィードバックする。このサイクルを回すことで診断率はおのずと向上する。保険で診断のついたところについてはこれ以上の研究コスト投入が不要になり、限られた研究費を研究すべき部分に重点的に充てられると西尾先生は考える。しかし、解析データ等の新規入力や更新、維持管理のためにかかる経費をどのように工面するかは、継続の壁だ。
難聴に関しては、今、原因診断のもう一歩先のフェーズに進んでいる。新しい治療法の開発につなげるというフェーズだ。「補聴器や人工内耳といった医療機器の性能が良いことに加え、投与法が難しいことが難聴に対する治療法開発が遅れてきた理由でした。内耳は骨に囲まれている組織で、しかも血管関門があるため経口投与や注射では薬が届かないのです」(西尾先生)。
しかし、アメリカでは昨年、 難聴の遺伝子治療でフェーズ1試験がスタートしているという。西尾先生は、全身に投与すると副作用が強くて過去のフェーズ試験で落ちてしまったような薬も、局所に用いるのであれば使える可能性があるのではと予想し、こういったドラッグ・ライブラリーをスクリーニングする評価システムの開発も手がけている。 「私たちの先行研究(参考資料9)などをもとに、もし適した薬を見つけることができたとして、その薬が効く遺伝子変異をもつ患者がどのくらいいるのかが、このデータストレージで確認できます。開発ターゲットを決める上でも必要不可欠な情報です」とデータストレージ利活用の展望を語った。
-
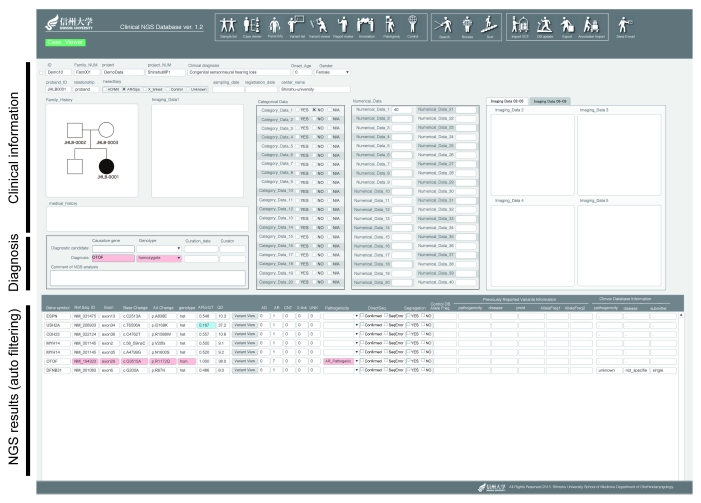 図3:難聴データストレージのケースビュアー画面。
図3:難聴データストレージのケースビュアー画面。
-
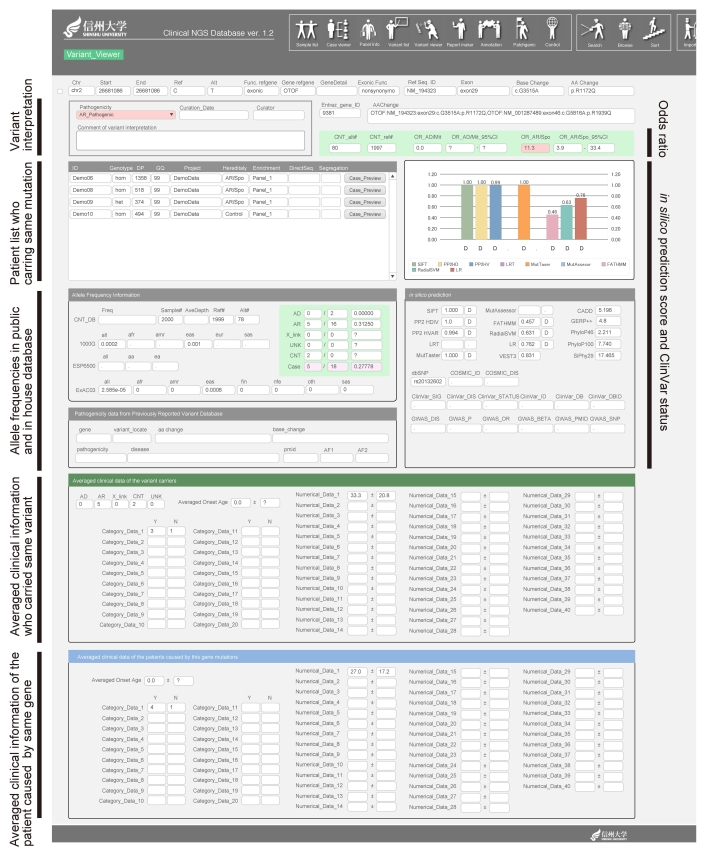 図4:難聴データストレージのバリアントビュアー画面。
図4:難聴データストレージのバリアントビュアー画面。
未来の難聴診断に求めるAIの力(ちから)

情報の機械学習によって、
AIが原因遺伝子のあたりを
付けられるようになるので
はと語る。
約8,000家系のデータを集められた要因は、検査方法の開発によるものだけではない。 宇佐美研の先天性難聴遺伝子変異のデータストレージには、クリニカルシークエンスのために得た患者のゲノム解析データだけでなく、北海道から沖縄まで全国・80医療施設から、信州大と共同研究契約を結ぶ検査会社を経由して"手渡し"で集まってきた手書きの情報も電子入力し、収められている。
「電子カルテ化が進んでいますから、今どき電子データで集めればいいのにとお思いになるでしょう。しかし、患者さんの詳細な臨床情報ですから、現時点では情報セキュリティの観点などからリスクが高いと考えていますし、実際の臨床現場では手書きで記入する方が(特に家系図などを記入する上では)むしろ簡便であるため、詳細な情報が得られていると考えています」臨床データがきちんと取られていて、ゲノム情報とつながっていることが、国際的な研究競争に勝てる強みになると西尾先生は強調した。
原因となる遺伝子変異が特定された場合には、データストレージのインターフェースの中でレポートを自動生成、打ち出せるようになっている。それら1件1件に対して、宇佐美先生がコメントを付けて返しているという。「近い将来、AIが遺伝子診断をアシストするAIアシステッド・ダイアグノーシスという形になるかもしれません。その精度にはおおいに期待するところです。でも、人がきちんと見て、最後に責任をもってコメントとサインをつけてお返しする、そのプロセスはすごく大事だと考えています。このような共同研究は人と人とのつながりで動いている部分がありますし、最終的には患者さんの治療に結び付けることが目的ですから」と未来の医療を展望した。
理学と医学の両方をバックグラウンドに持ち、Wet・Dry両方を担当する西尾先生。宇佐美先生とともに、ターゲット・リシークエンスのライブラリ作成、NGS解析、得られたデータの情報解析(アノテーション)、見出された変異の病原性の判断(インタープリテーション)、診断委員会のボードメンバー、データベースの構築・運用、担当医向けの結果返却報告書の作成などプロジェクト全体の運営に携わる。「NGSの臨床応用により遺伝子解析に関しては大幅な進展を見せており、従来であればできなかったようなさまざまな研究が行えるようになってきています。NGS解析は、まさに情報学と生物学の融合分野。Wet研究とDry研究の両方を学ぶことがこれからの研究者には必要になるでしょう。Dryの研究者にとっては、Wet解析を学ぶことを通じて、どのような部分でヒューマンエラーが起こるか、データのばらつきが生じるかなどを学ぶとともに、実際の臨床でどのような検査が行われ、その結果にどのような意義があるかを知ることが必要。一方、Wetの研究者にとっても、研究ツールの1つとしてDry解析を用いる技術やDry解析の原理などの理解が求められるでしょう 」と若手研究者に向けての期待するとともに、そういった人材を育成しながらビッグデータ活用で研究-医療還元の回転サイクルを加速させる姿を強く望んでいる。
インタビュー動画
- コラム研究者紹介(youtube動画)
推薦論文
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
The Clinical Next-Generation Sequencing Database: A Tool for the Unified Management of Clinical Information and Genetic Variants to Accelerate Variant Pathogenicity Classification.
著者名 Nishio SY, Usami SI.
雑誌名 Hum Mutat.
号、発行年 2017 Mar;38(3):252-259.
研究者経歴
岐阜県生まれ。1998年に信州大学理学部生物学科卒業。2008年に信州大学大学院工学系研究科地球環境システム科学専攻 単位取得退学。 2008年9月に信州大学医学部附属病院耳鼻咽喉科 研究員として着任。(財)長寿科学振興財団・リサーチレジデント、信州大学医学部耳鼻咽喉科 助教を経て、現職。
掲載日 平成30年10月11日
最終更新日 令和2年3月30日